
Pathologie digestive - b) Tube digestif
Plan : - Item 271 – Reflux gastro-œsophagien chez le nourrisson, chez l’enfant et chez l’adulte. Hernie hiatale
- Item 272 – Ulcère gastrique et duodénal. Gastrite
- Item 273 – Dysphagie
- Item 282 – Maladies inflammatoires chroniques de l’intestin (MICI) chez l’adulte
- Item 285 – Diarrhée chronique chez l’adulte et l’enfant
- Item 305 – Tumeurs de l’œsophage
- Item 301 – Tumeurs du côlon et du rectum
- Item 303 – Tumeurs de l’estomac
b) Tube digestif
Item 271 – Reflux gastro-œsophagien chez le nourrisson, chez l’enfant et chez l’adulte. Hernie hiatale
Auteure : Camille Boulagnon-Rombi
I. Prérequis : histologie de l’œsophage
II. Prévalence et facteurs de risque du reflux gastro-œsophagien
III. Diagnostic du reflux gastro-œsophagien
IV. Principales complications du reflux gastro-œsophagien
V. Surveillance
Hiérarchisation des connaissances Tableau 1
{kind=link}
I -Prérequis : histologie de l’œsophage
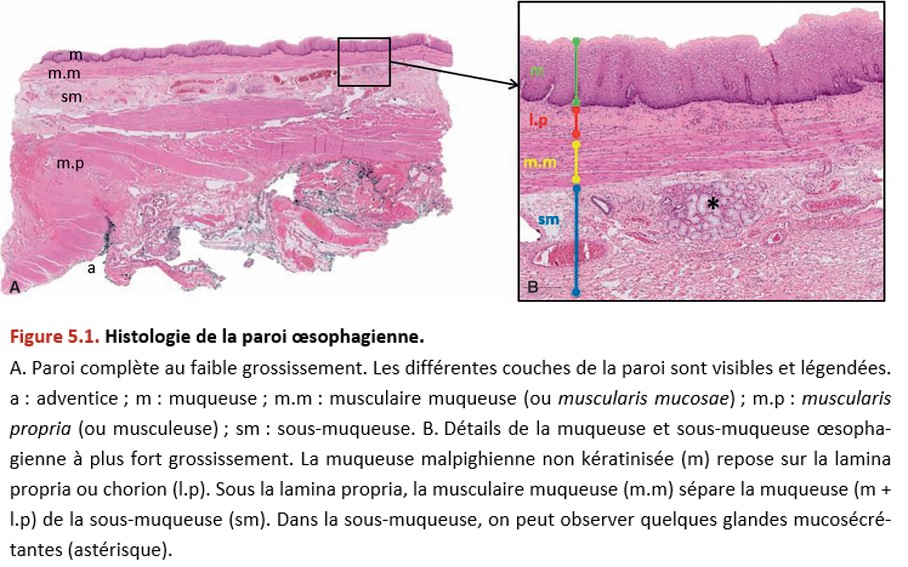
La paroi œsophagienne est constituée histologiquement (Figure 5.1) :
{kind=link}
• d’une muqueuse composée :
– de l’épithélium, qui est de type malpighien non kératinisé. Il repose sur une membrane basale,
– du chorion (ou lamina propria), qui est le tissu conjonctif de soutien,
– de la musculaire muqueuse (ou muscularis mucosae), qui est une fine couche de cellules musculaires lisses ;
• d’une sous-muqueuse contenant quelques glandes ;
• d’une musculeuse (deux couches musculaires lisses) ou muscularis propria ;
• d’une adventice (attention : au niveau de l’œsophage, il n’y a pas de séreuse, on ne parle donc pas de sous-séreuse).
L’œsophage se continue par l’estomac (cardia). La jonction œsogastrique se voit en endoscopie, la muqueuse malpighienne a un aspect blanc na-cré et la muqueuse gastrique cardiale (glandulaire) a un aspect rosé.
II. Prévalence et facteurs de risque du reflux gastro-œsophagien
Le RGO est majoritairement bénin. Il faut distinguer le RGO « physiologique » et le RGO « pathologique ». En effet, la définition du RGO est fonctionnelle et implique que les remontées ne sont pas toutes symptomatiques, ni toutes acides.
Le RGO « pathologique » est caractérisé par des symptômes gênants (principalement le pyrosis) et/ou des complications. Dans ce texte, le terme RGO est à comprendre comme un RGO pathologique.
Le RGO est fréquent. Environ 10 % de la population générale rapporte un épisode de pyrosis par semaine et 2 à 5 % un pyrosis quotidien.
L’obésité est le principal facteur de risque de RGO. D’autres facteurs de risque peuvent favoriser le RGO : hernie hiatale, grossesse, alcool, certaines chirurgies bariatriques et certains médicaments.
III. Diagnostic du reflux gastro-œsophagien
Lors de l’EOGD (endoscopie œso-gastro-duodénale), les biopsies sont systématiques :
• s’il existe une sténose (recherche de cancer) ;
• s’il y a un aspect endoscopique évocateur d’endobrachyœsophage (EBO) ;
• en cas de suspicion d’une tumeur ou d’une autre cause d’œsophagite.
Les ulcérations peptiques aiguës (liées aux remontées acides) ne sont pas biopsiées.
Les ulcérations ou ulcères résistants au traitement sont en général biopsiés.
• le diagnostic d’EBO ;
• le diagnostic d’une éventuelle lésion tumorale ulcérée ;
• en cas d’œsophagite : l’identification éventuelle de certains agents pathogènes à traitement spécifique (œsophagite à Candida/œsophagite herpétique).
IV. Principales complications du reflux gastro-œsophagien
A. Endobrachyœsophage
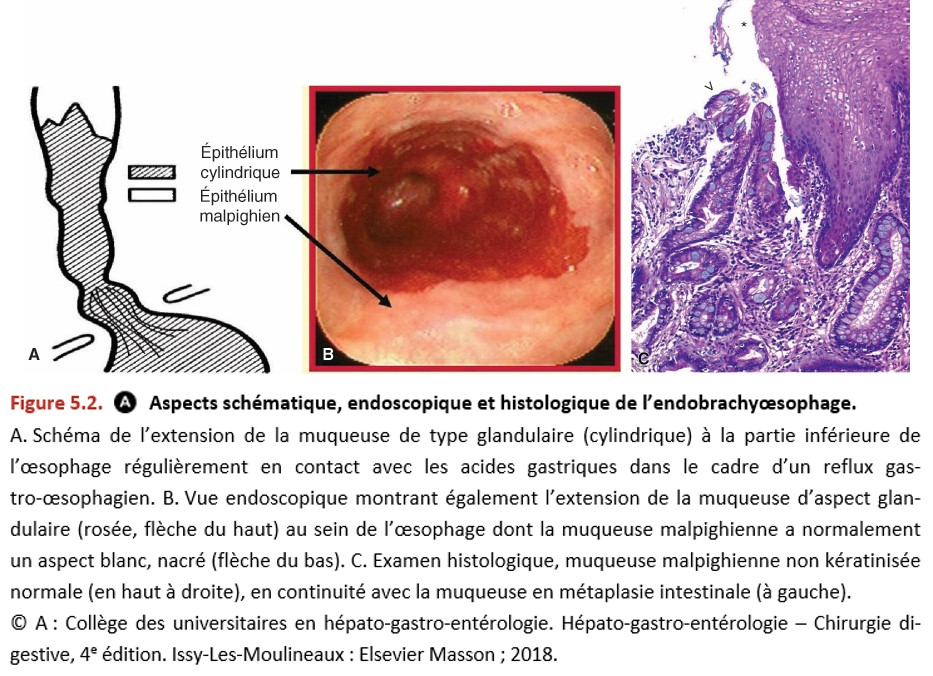
L’EBO, aussi appelé œsophage de Barrett, correspond à une transformation de l’épithélium malpighien de l’œsophage en un épithélium glandulaire, le plus souvent de type intestinal (métaplasie intestinale). Le diagnostic est endoscopique et confirmé par l’examen anatomopathologique de biopsies de la muqueuse œsophagienne anormale (Figure 5.2). L’aspect endoscopique est celui d’une « ascension » de la ligne Z de façon circonférentielle ou en languette, avec un décalage de plus de 1 cm de hauteur par rapport au sommet des plis gastriques. Au niveau histologique, la muqueuse malpighienne non kératinisée normale de l’œsophage est remplacée par une muqueuse glandulaire avec métaplasie intestinale (également appelée « muqueuse spécialisée ») sur des biopsies de topographie œsophagienne (hors cardia).
{kind=link}
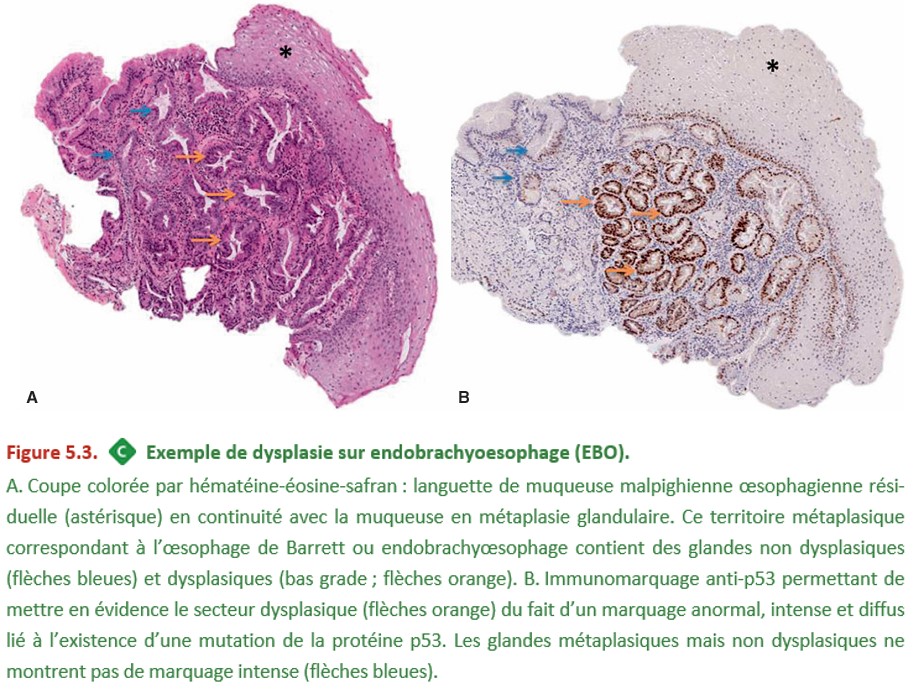
Des marquages immunohistochimiques (expression anormale de p53 ou des cellules en cycle exprimant Ki-67 au sein de l’épithélium) peuvent aider au diagnostic (Figure 5.3). Lorsque le diagnostic de dysplasie est incertain (douteux), on utilise les termes suivants : « indéfini pour la dysplasie ». En cas de dysplasie de haut grade (sévère), il est recommandé que le diagnostic soit confirmé par la relecture des biopsies par un deuxième pathologiste (expert).
{kind=link}
Terminologie utilisée dans les comptes rendus
•
• Métaplasie intestinale (ou muqueuse de Barrett/EBO/muqueuse spécialisée) indéfinie pour la dysplasie (dysplasie douteuse).
• Métaplasie intestinale (ou muqueuse de Barrett/EBO/muqueuse spécialisée) avec dysplasie de bas grade.
• Métaplasie intestinale (ou muqueuse de Barrett/EBO/muqueuse spécialisée) avec dysplasie de haut grade.
• Adénocarcinome.
Les caractéristiques endoscopiques et histologiques suivantes conditionnent les modalités de surveillance endoscopique et de traitement :
• hauteur de l’atteinte en endoscopie (< ou ≥ 3 cm) ;
• présence d’une dysplasie de bas ou de haut de grade.
B. Adénocarcinome de l’œsophage
V. Surveillance
La surveillance repose toujours sur la fibroscopie avec biopsies multiples selon le protocole de Seattle :
• pour les EBO < 3 cm : 2 à 4 biopsies par cm d’EBO ;
• pour les EBO ≥ 3 cm : biopsies des quatre quadrants tous les 2 cm selon le protocole de Seattle.
La conduite à tenir en fonction des résultats anatomopathologiques est indiquée dans le Tableau 5.1
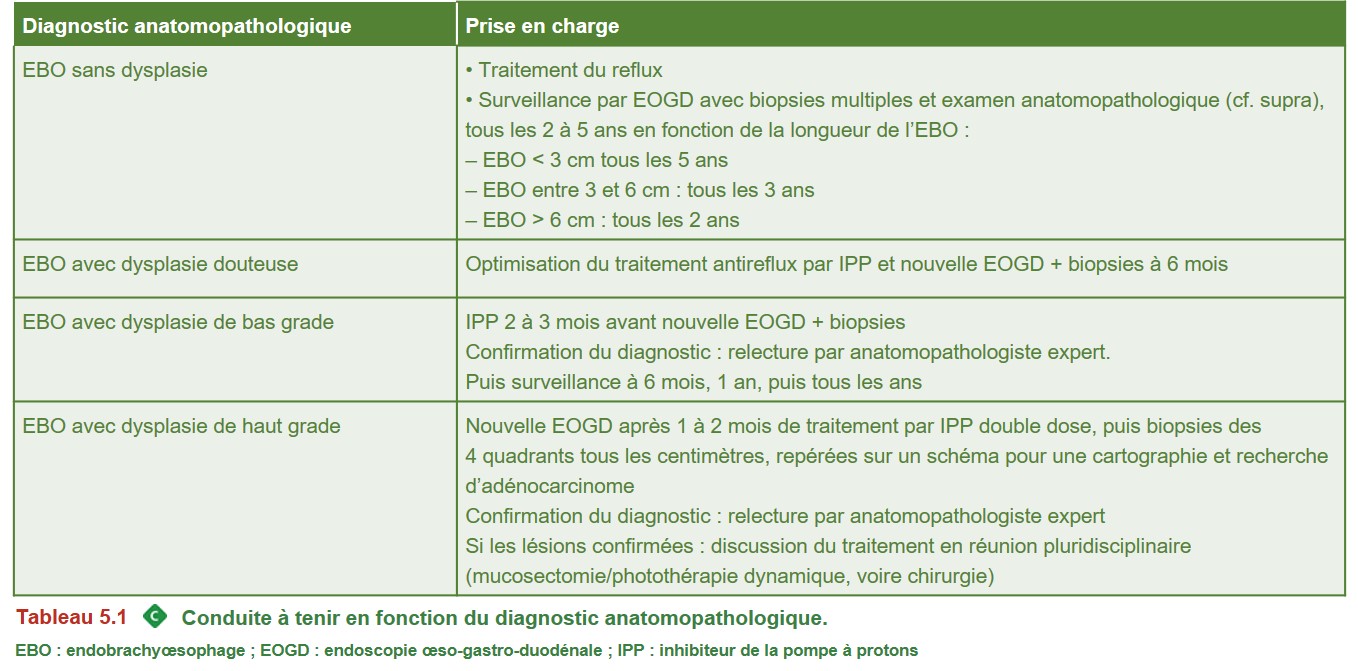{kind=link}
EBO : endobrachyœsophage ; EOGD : endoscopie œso-gastro-duodénale ; IPP : inhibiteur de la pompe à protons
Points clés
• L’examen anatomopathologique des biopsies œsophagiennes dans un contexte de RGO a comme but principal :
– le diagnostic d’EBO ;
– le diagnostic d’une éventuelle lésion tumorale ;
– l’identification possible de certains agents pathogènes à traitement spécifique (œsophagite à Candi-da/œsophagite herpétique).
• L’EBO ou œsophage de Barrett est défini par la présence d’une muqueuse glandulaire métaplasique (intestinale) dans l’œsophage, visible en endoscopie. Il s’agit d’un diagnostic conjoint endoscopique et histologique.
• L’EBO est secondaire à un RGO ancien et sévère ; la réalisation de biopsies est indispensable pour confirmer le diagnostic
• L’EBO est une lésion précancéreuse qui expose à un risque de cancer (adénocarcinome), par la sé-quence métaplasie sans dysplasie/dysplasie de grade croissant/adénocarcinome.
• La dysplasie est diagnostiquée sur les biopsies.
Item 272 – Ulcère gastrique et duodénal. Gastrite
Auteure : Camille Boulagnon-Rombi
I. Prérequis : histologie de la paroi gastrique
II. Ulcère gastrique et duodénal
III. Gastrites
Hiérarchisation des connaissances Tableau 2
{kind=link}
I. Prérequis : histologie de la paroi gastrique
L’estomac est composé histologiquement de trois régions :
• le cardia : portion proximale de l’estomac en continuité avec l’œsophage :
• le fundus, qui fait suite au cardia et qui est la plus grande partie de l’estomac ;
• l’antre à la partie inférieure de l’estomac juste avant la région pylorique et le duodénum.
La paroi gastrique est constituée de :
• la muqueuse (épithélium de surface + chorion + musculaire muqueuse) ;
• la sous-muqueuse ;
• la musculeuse ;
• la sous-séreuse ;
• la séreuse.
L’estomac étant un organe intrapéritonéal, il est recouvert de péritoine : la séreuse. Le tissu con-jonctif situé entre la musculeuse et la séreuse est donc appelé la sous-séreuse.
Il existe deux principaux types de muqueuse gastrique (Figure 5.4) :
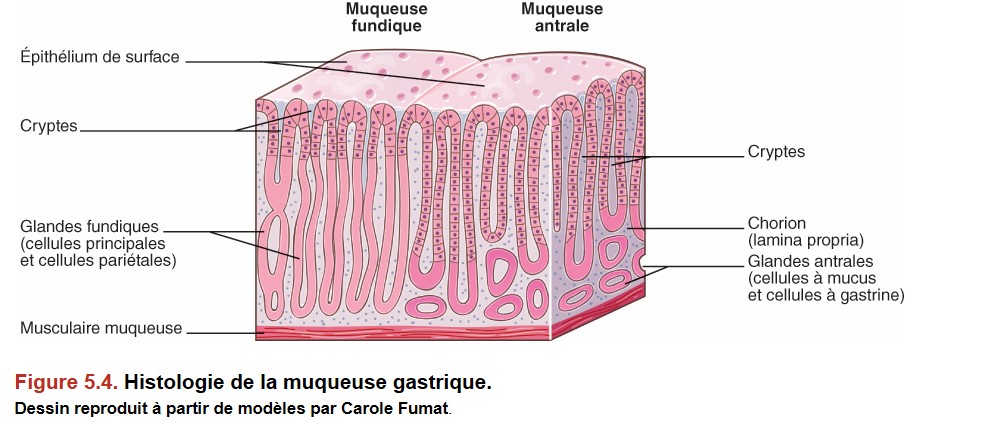{kind=link}
• la muqueuse de type fundique située au niveau de la grosse tubérosité et du corps de l’estomac ;
• la muqueuse de type antral ou pylorique au niveau de l’antre.
La transition entre les deux types de muqueuse ne se voit pas en endoscopie.
L’épithélium de surface et celui des cryptes sont le même dans tout l’estomac : il s’agit d’un épithélium à pôle muqueux « fermé » (rôle surtout de protection).
Les glandes gastriques sont différentes dans le fundus et l’antre.
Les glandes fundiques sont faites essentiellement de cellules :
• pariétales (sécrétion d’HCl et de facteur intrinsèque) ;
• principales (sécrétion de pepsinogène).
Il existe également des cellules endocrines produisant de l’histamine : cellules « ECL » (enterochromaffin like).
Les glandes antrales sont constituées principalement de cellules :
• mucosécrétantes ;
• endocrines à gastrine (la gastrine est une hormone stimulant la synthèse d’HCl par les glandes fundiques et stimulant la prolifération des cellules ECL du fundus).
II. Ulcère gastrique et duodénal
A. Définition et facteurs de risque
L’ulcère gastrique peut correspondre à une complication de l’infection par Helicobacter pylori (environ 70 % des cas) mais peut aussi être lié à :
• la présence d’une lésion tumorale +++ ;
• une prise d’AINS ;
• une maladie de Crohn ;
• d’autres causes rares (syndrome de Zollinger-Ellison, réanimation, etc.).
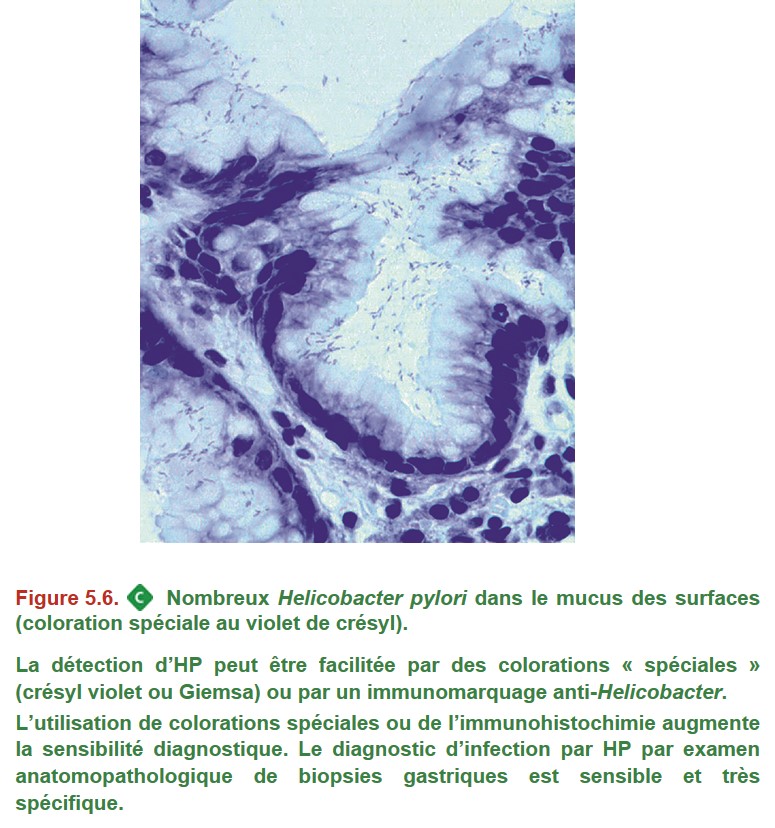
B. Helicobacter pylori (HP)
Sa prévalence est élevée dans les pays en voie de développement et dans les populations les plus âgées, son incidence est en décroissance depuis 30 ans. HP s’installe préférentiellement dans l’antre, mais il peut infecter le fundus. L’infection antrale, active, peu atrophique entraîne une hypergastrinémie avec hyperchlorhydrie qui pourra provoquer des lésions de bulbite et un ulcère duodénal. Puis une atrophie de la muqueuse antrale apparaît chez certains individus, avec achlorhydrie et installation d’HP dans le fundus. La gastrite atrophique devient diffuse et fragilise la muqueuse gastrique avec risque surtout d’ulcère gastrique. HP est donc à l’origine d’une gastrite chronique avec atrophie, métaplasie puis parfois dysplasie, avec un risque accru d’adénocarcinome de l’estomac. Par ailleurs, le lymphome à petites cellules de type MALT (mucosae-associated lymphoid tissue) est quasiment toujours associé à une infection par HP (> 90 %).
Tout syndrome ulcéreux nécessite la réalisation d’une EOGD.
•
• Si l’endoscopie montre un ulcère gastrique, il faut faire des biopsies gastriques avec examen anatomopathologique pour faire le diagnostic de gastrite à HP, et des biopsies multiples de l’ulcère gastrique pour rechercher une lésion tumorale (adénocarcinome, voire lymphome) : 8 à 10 biopsies intéressant toute la circonférence de la lésion sur les berges de l’ulcère.
Après traitement, une fibroscopie de contrôle doit être réalisée avec des biopsies de la zone ulcérée résiduelle ou de la zone cicatricielle, et des biopsies gastriques antrales et fundiques pour recherche d’HP (échec de l’éradication dans environ 30 %).
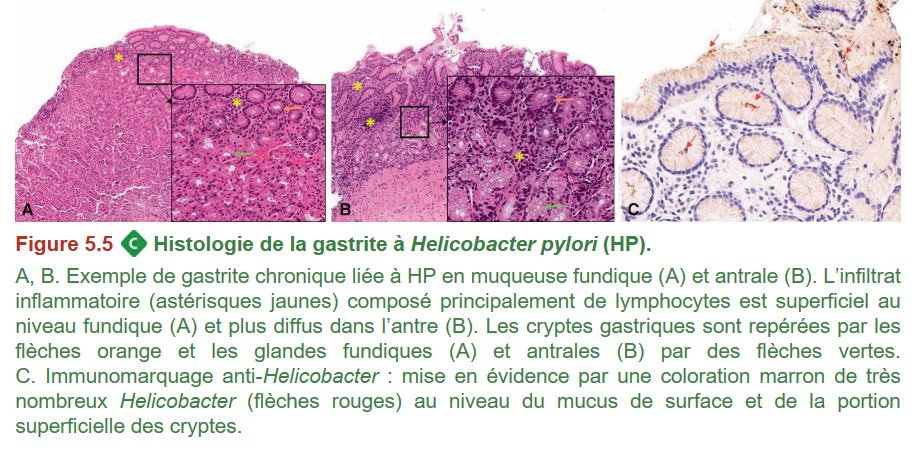{kind=link}
{kind=link}
C. Complications
•
• Perforation
• Sténose
• Pour l’ulcère gastrique uniquement : transformation en cancer (adénocarcinome)
Les cancers gastriques peuvent avoir l’aspect d’un ulcère bénin, ce qui justifie le contrôle de leur cicatrisation en endoscopie (avec biopsies multiples des berges de l’ulcère).
Les ulcères du duodénum n’évoluent jamais vers un cancer.
III. Gastrites
A. Définition et principaux types
La gastrite correspond à une inflammation aiguë ou chronique de la muqueuse gastrique. Elle est diagnostiquée en anatomopathologie sur les biopsies pratiquées lors d’une EOGD.
La définition et le diagnostic de gastrite sont histologiques.
Il s’agit de lésions inflammatoires de la muqueuse gastrique.
Il existe une très mauvaise corrélation entre la clinique, l’aspect endoscopique et l’histologie. Le terme de gastrite ne doit pas être utilisé pour décrire des symptômes ou un aspect endoscopique.
Les atteintes par des pathologies non inflammatoires (par exemple AINS, alcool, bile, hypertension portale) de la muqueuse gastrique sont appelées des gastropathies.
• la cause (cf. infra) ;
• la localisation des lésions dans le corps gastrique : antrale, fundique, pangastrique ;
• le type de lésions anatomopathologiques : inflammation chronique et ou active, atrophie glandulaire, métaplasie, présence ou non de pathogène.
Exemple :
• gastrite auto-immune = muqueuse fundique ;
• gastrite à Helicobacter pylori = muqueuse antrale ou pangastrique.
Pour porter un diagnostic de gastrite, il est recommandé de faire des biopsies en muqueuse antrale et fundique.
B. Causes
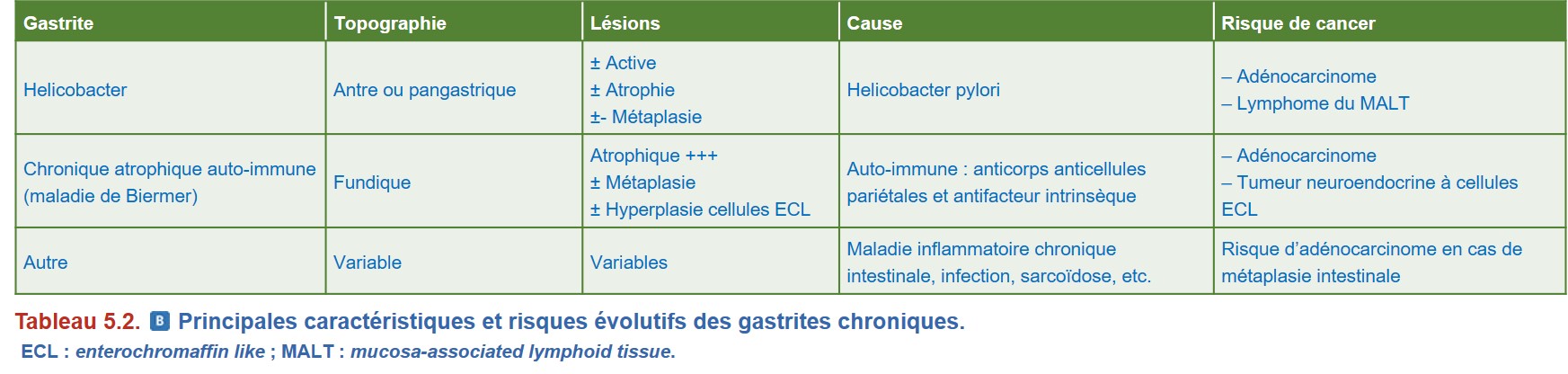
La cause la plus fréquente est la gastrite liée à HP (Tableau 5.2)
{kind=link}
Les autres causes plus rares sont :
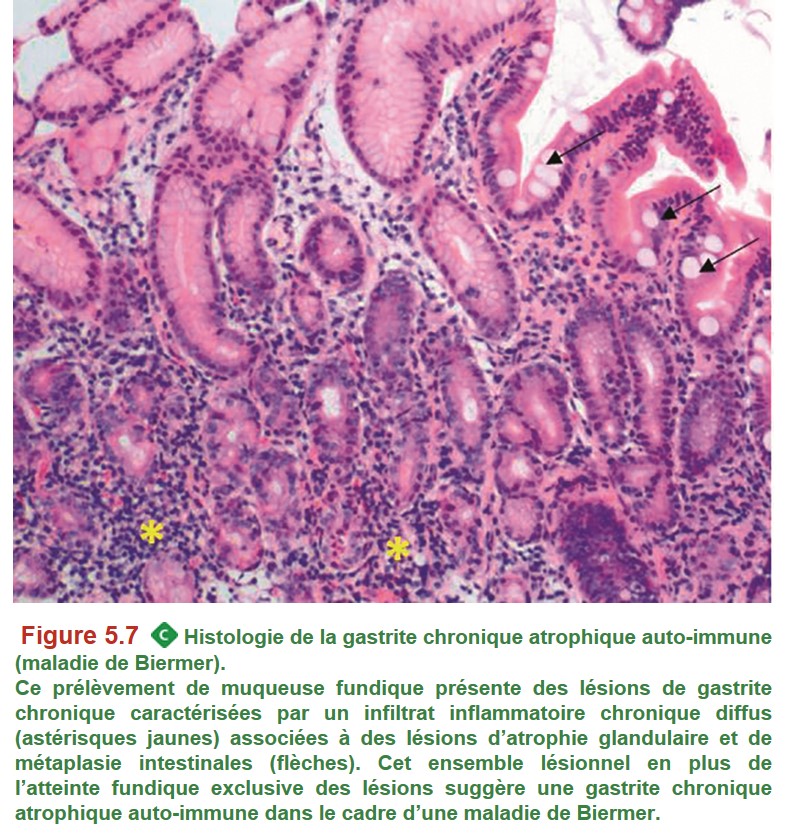
• la gastrite chronique atrophique auto-immune ou « maladie de Biermer ». Relativement fréquente chez les sujets âgés, elle peut survenir plus tôt, notamment dans un contexte familial ou personnel d’auto-immunité. C’est une maladie auto-immune avec présence d’anticorps anticellules pariétales (cellules présentes dans les glandes fundiques synthétisant l’HCl et le facteur intrinsèque) et antifacteur intrinsèque.
{kind=link}
•
•
• la gastrite granulomateuse. Elle correspond à la présence de granulomes épithélioïde et gi-gantocellulaire au sein de la muqueuse gastrique. Elle peut être associée à une maladie de Crohn, une sarcoïdose, la présence de corps étrangers, certains déficits immunitaires innés rares, etc. ;
• la gastrite aiguë infectieuse, qui peut être virale (à herpèsvirus chez le sujet immunodéficient) ou phlegmoneuse d’origine bactérienne ; elle est rare.
C. Indications et place des biopsies pour le diagnostic
L’indication des biopsies dépend de la clinique et de l’aspect endoscopique de la muqueuse gastrique :
• en présence de lésion endoscopique : affirmer le diagnostic de gastrite et en rechercher la cause ;
• en présence de signes cliniques (épigastralgies) et d’endoscopie normale : biopsies à la recherche d’une pathologie spécifique.
Par exemple, en cas de carence en vitamine B12 et d’anémie macrocytaire, des biopsies endoscopiques gastriques sont indiquées à la recherche d’une atrophie. De même, en cas d’antécédent familial de cancer gastrique, ou en cas de douleurs abdominales chroniques, des biopsies endoscopiques gastriques sont indiquées afin de rechercher une gastrite à HP.
D. Caractérisation anatomopathologique
• le type de muqueuse gastrique atteint (antrale, fundique, les deux) ;
• l’importance de l’inflammation lymphocytaire et plasmocytaire ;
• l’existence ou non d’une activité définie par la présence de polynucléaires neutrophiles et son intensité ;
• l’existence ou non d’une atrophie glandulaire et son intensité ;
• l’existence ou non d’une métaplasie intestinale et son intensité (préciser alors s’il y a de la dysplasie associée) ;
• la présence ou non d’HP.
Points clés
• La réalisation d’une EOGD avec biopsies gastriques est à pratiquer devant tout syndrome ulcéreux.
• La recherche d’HP est systématique devant tout UGD, réalisée sur les biopsies gastriques pratiquées pendant l’endoscopie diagnostique.
• Un contrôle de la cicatrisation des ulcères gastriques en endoscopie (avec biopsies) est nécessaire mais pas celui de la cicatrisation des ulcères duodénaux.
• Le diagnostic de gastrite est histologique.
• Il requiert des biopsies du fundus et de l’antre.
• La gastrite à HP est localisée préférentiellement dans l’antre.
• La gastrite chronique atrophique auto-immune (Biermer) est localisée exclusivement dans le fundus.
• Les gastrites chroniques peuvent favoriser la survenue d’adénocarcinomes gastriques par le biais de la voie métaplasie, dysplasie de grades croissant, cancer.
Item 273 – Dysphagie
Auteure : Camille Boulagnon-Rombi
I. Prérequis
II. Définition et explorations
III. Les principales étiologies
Hiérarchisation des connaissances Tableau 3
{kind=link}
I. Prérequis
Histologie de l’œsophage (cf. chapitre supra item 271 – Reflux gastro-œsophagien chez le nourrisson, chez l’enfant et chez l’adulte. Hernie hiatale).
II. Définition et explorations
Selon le siège de la dysphagie, on distingue :
• les dysphagies cervicopharyngées (ou dysphagies hautes ; souvent d’origine neurologique ou ORL) ;
• les dysphagies œsophagiennes.
Il est possible de différencier les dysphagies selon leur mécanisme :
• dysphagie lésionnelle (liée à une lésion organique œsophagienne ou extrinsèque) ;
• dysphagie fonctionnelle, sans lésion identifiée.
Le but de l’exploration est de déterminer s’il s’agit d’une dysphagie lésionnelle ou fonctionnelle.
Toute dysphagie est lésionnelle (cancer de l’œsophage ou oropharyngé) jusqu’à preuve du contraire.
Toute dysphagie de l’adulte nécessite la réalisation d’une EOGD avec biopsies multiples de la lésion et examen anatomopathologique. Les biopsies intéressent la partie superficielle de la paroi œsophagienne, c’est-à-dire la muqueuse. L’endoscopie permet :
• d’affirmer ou infirmer le caractère lésionnel de la dysphagie ;
• la réalisation de biopsies ciblées en cas de lésion œsophagienne ;
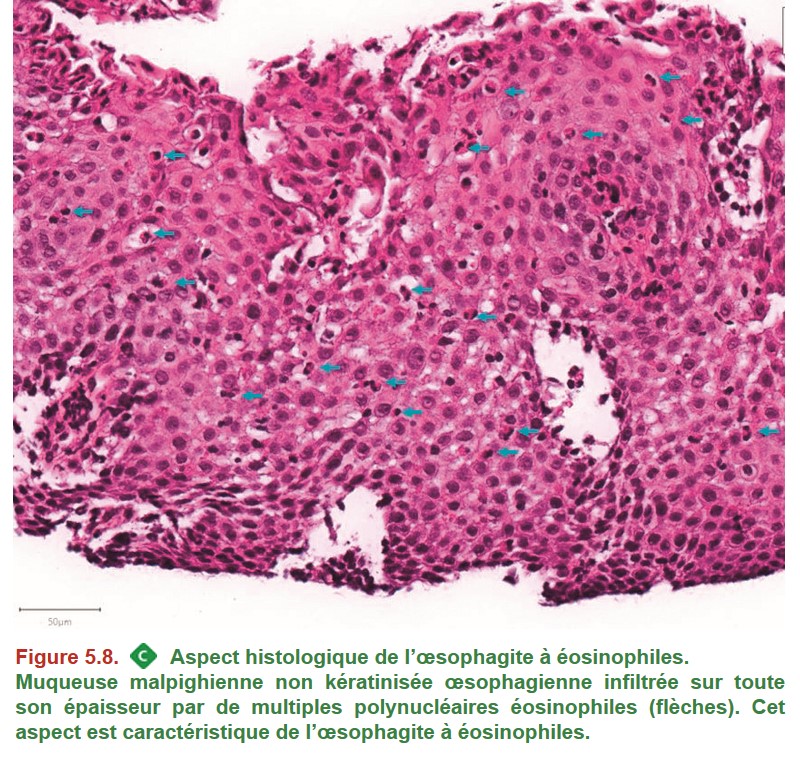
• la réalisation de biopsies œsophagiennes systématiques en l’absence de lésion visible (recherche d’œsophagite à éosinophiles).
III. Principales étiologies
A. Dysphagies œsophagiennes lésionnelles
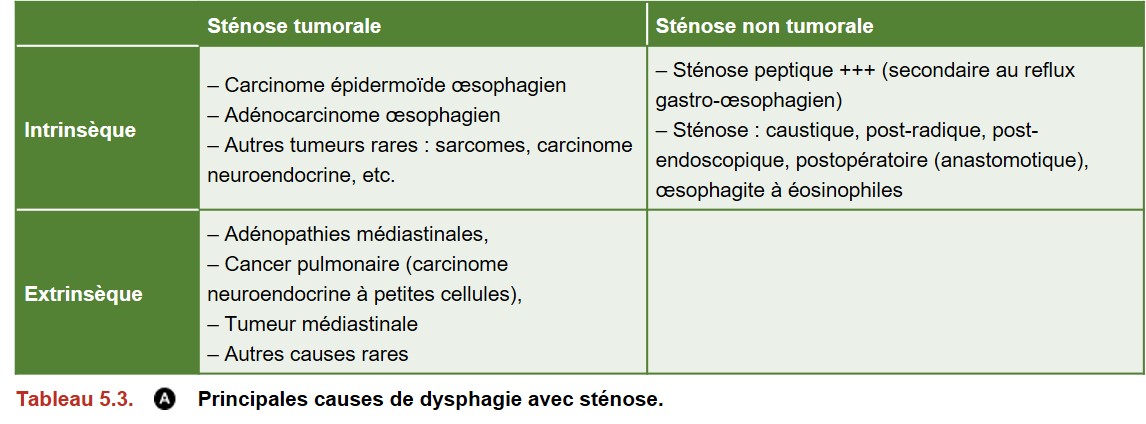
Une sténose de l’œsophage doit faire évoquer une lésion tumorale ; les principales causes de dysphagie avec sténose sont présentées dans le tableau Tableau 5.3
{kind=link}
Les principales causes de dysphagie non tumorales sont :
• l’œsophagite sévère : peptique, caustique, post-radique, médicamenteuse, infectieuse (myco-tique, virale, etc.). Elle peut parfois s’accompagner d’une sténose ;
• les diverticules œsophagiens :
– cervical, dit de Zenker (diverticule de la face postérieure de l’œsophage, à la jonction pharyngo-œsophagienne),
– du tiers inférieur de l’œsophage, dit épiphrénique, souvent associé aux troubles moteurs œsophagiens ;
•
{kind=link}
B. Dysphagies pharyngolaryngées lésionnelles sténosantes
•
• Causes extrinsèques : goitre thyroïdien compressif, anévrisme de l’aorte thoracique, tumeurs du médiastin.
• Autres causes œsophagiennes hautes : séquelles d’ingestion de caustique, syndrome de Plummer-Vinson.
Points clés
Toute dysphagie est lésionnelle (cancer de l’œsophage ou oropharyngé) jusqu’à preuve du contraire.
• L’examen d’exploration à réaliser en 1re intention est une EOGD.
• Les dysphagies lésionnelles avec sténose sont majoritairement d’origine tumorale.
• Des biopsies ciblées doivent être réalisées en cas de lésions visibles à l’EOGD.
• Des biopsies œsophagiennes systématiques doivent être réalisées en l’absence de lésion visible (recherche d’œsophagite à éosinophiles).
Item 282 – Maladies inflammatoires chroniques de l’intestin (MICI) chez l’adulte
Auteure : Camille Boulagnon-Rombi
I. Prérequis : histologie de l’intestin grêle et du côlon
II. Définitions et caractéristiques anatomopathologiques
III. Diagnostic
IV. Complications
Hiérarchisation des connaissances Tableau 4
{kind=link}
I. Prérequis : histologie de l’intestin grêle, du côlon et du rectum
Comme pour l’estomac (cf. supra item 272 – Ulcère gastrique et duodénal. Gastrite), la paroi intestinale grêle et la paroi colique sont constituées de :
– la muqueuse (épithélium + chorion + musculaire muqueuse) ;
– la sous-muqueuse : tissu conjonctif vascularisé, réseau de nerfs sympathiques. Au niveau du duodénum, la sous-muqueuse contient des glandes de Brünner (secrètent un mucus alcalin visant à protéger la muqueuse duodénale de l’acidité gastrique) ;
– la musculeuse : elle comprend une couche circulaire interne et une couche longitudinale externe, constituées de cellules musculaires lisses. Entre ces deux couches, on observe des plexus nerveux, les plexus myentériques (d’Auerbach), responsables de l’innervation végétative du tube digestif ;
– la sous-séreuse : tissu adipeux et vascularisé contenant des ganglions lymphatiques ;
– la séreuse.
Au niveau du moyen et du bas rectum, il n’existe pas à proprement parler de séreuse (pas de péritoine). Le tissu adipeux périrectal au-delà de la musculeuse est nommé mésorectum.
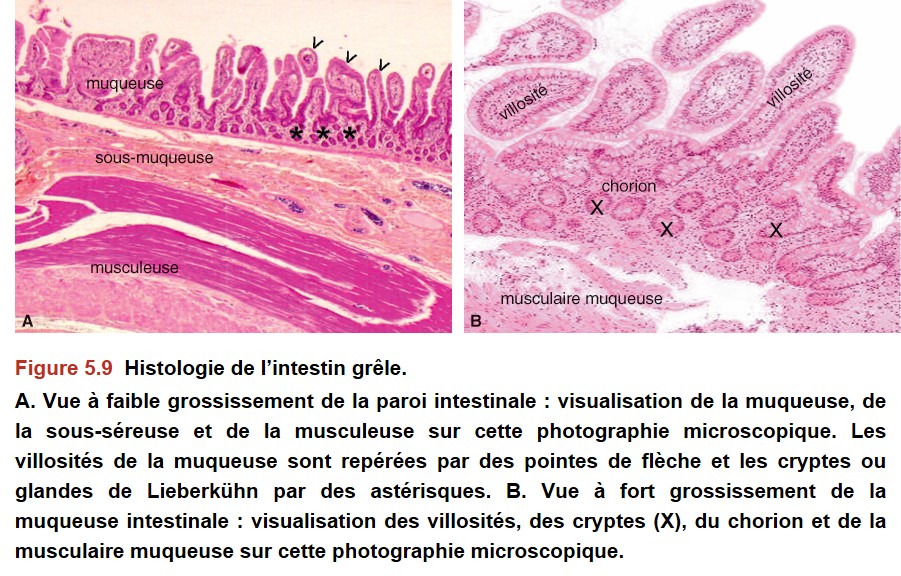
La muqueuse intestinale (duodénum, jéjunum, iléon) est constituée de villosités et de cryptes (Figure 5.9).
{kind=link}
La muqueuse colique et rectale est constituée d’un épithélium de surface et de cryptes ou glandes dites « de Lieberkühn » (Figure 5.10).
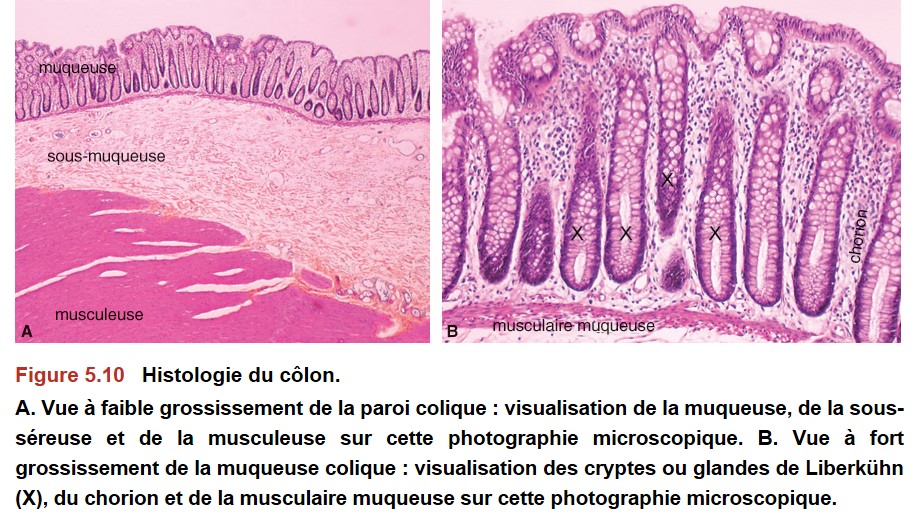{kind=link}
L’épithélium de surface, celui des villosités et des cryptes est composé d’entérocytes et de cel-lules caliciformes (cellules mucosécrétantes à pôle muqueux ouvert en « gobelet »).
La proportion de ces cellules varie en fonction de la localisation dans le tube digestif.
II. Définitions et caractéristiques anatomopathologiques
• maladie de Crohn (MC) ;
• rectocolite hémorragique (RCH).
Dans moins de 10 % des cas, le diagnostic hésite entre ces deux entités, c’est une colite indéterminée. Les MICI prédisposent au cancer colorectal en cas d’atteinte colique (risque plus élevé pour la RCH).
Elles touchent plutôt les sujets jeunes (enfants, adultes jeunes) avec un second pic autour de 60 ans dans la RCH.
• ulcérations ;
• raréfaction des glandes ;
• infiltrat inflammatoire lymphocytaire et plasmocytaire.
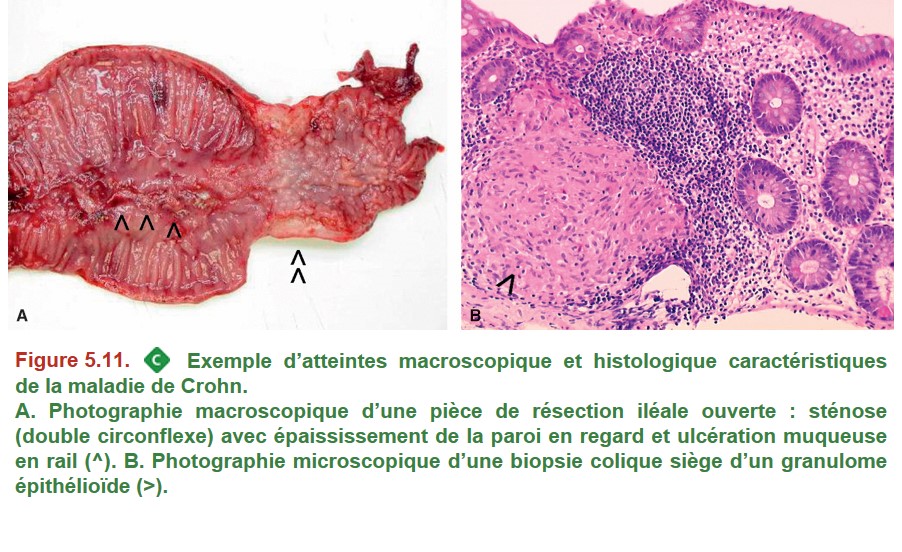
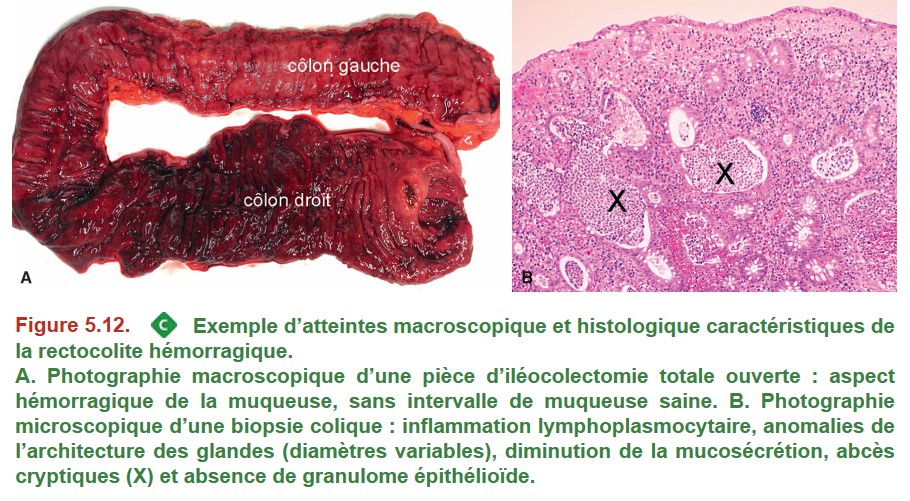
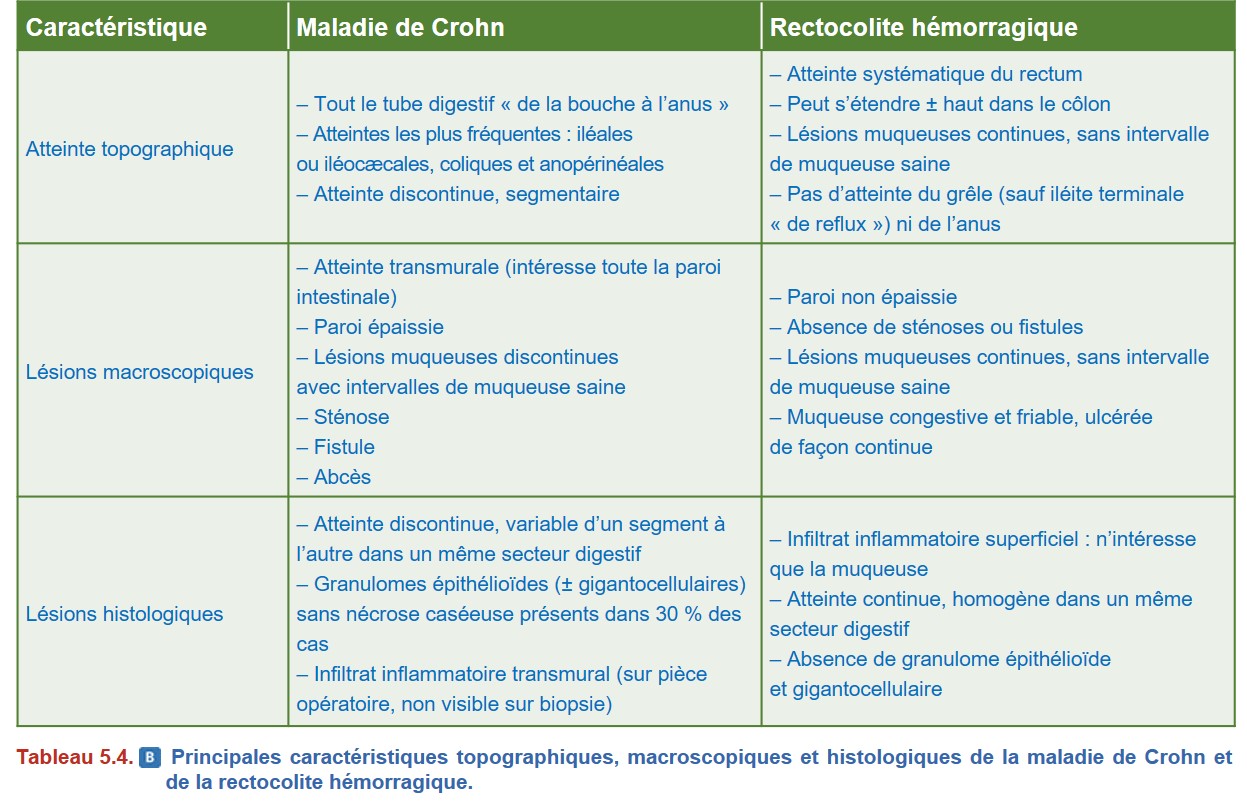
Cependant, la topographie des lésions ainsi que leur aspect macroscopique et microscopique peuvent orienter le diagnostic vers l’une ou l’autre des MICI. Les principales caractéristiques topographiques, macroscopiques et microscopiques de la MC et de la RCH sont résumées dans le Tableau 5.4 et illustrées dans la Figure 5.11 et la Figure 5.12.
{kind=link}
{kind=link}
{kind=link}
III. Diagnostic
Les manifestations cliniques dépendent du type de MICI, de la localisation, de l’intensité et de l’ancienneté des lésions. Il peut exister des manifestations extra-intestinales et la maladie peut être révélée par une complication. Dans tous les cas, une iléocoloscopie doit être réalisée afin de mettre en évidence les lésions muqueuses et de faire des biopsies pour examen anatomopathologique. En cas de suspicion de MC, une EOGD est également recommandée.
• des lésions inflammatoires aiguës telles que les ulcérations, l’inflammation à polynucléaires neutrophiles dans les glandes ou la lamina propria (chorion) ;
• des lésions inflammatoires chroniques telles que l’augmentation de l’infiltrat inflammatoire chronique (lymphocytes, plasmocytes) et les anomalies de l’architecture des glandes.
Les autres éléments histologiques plus discriminants, sans être spécifiques, qui permettent le plus souvent de classer la MICI, sont détaillés dans le Tableau 5.4.
{kind=link}
• multiples ;
• étagées (critères topographiques très importants pour le diagnostic) ;
• en muqueuse d’aspect pathologique et sain.
En effet, les biopsies en muqueuse d’aspect endoscopique normal peuvent montrer des lésions microscopiques (inflammation chronique, granulomes) aidant au diagnostic.
Remarque : Les biopsies sont des prélèvements superficiels et n’intéressent que la muqueuse et éventuellement la partie superficielle de la sous-muqueuse.
L’étude de toute la paroi digestive à la recherche de l’ensemble des caractéristiques permettant de classer la MICI se fait sur une pièce opératoire lorsqu’une résection chirurgicale est pratiquée au cours de l’évolution de la maladie.
IV. Complications
• L’inflammation chronique favorise le développement d’une fibrose intestinale qui peut évoluer vers une sténose. Cela est le plus souvent observé au niveau du grêle dans la MC.
• L’inflammation chronique est également un facteur de carcinogenèse (séquence dysplasie de grade croissant/adénocarcinome). Le risque de cancer colorectal est augmenté dans la RCH en particulier, et aussi la MC.
• Le risque de dysplasie augmente avec la durée de la maladie, son étendue et la persistance d’une inflammation active. Un dépistage de la dysplasie par endoscopie + biopsies est donc nécessaire en fonction de la durée d’évolution de la maladie et de son étendue. Dans la RCH, la présence d’une cholangite sclérosante primitive associée augmente de façon très importante le risque de cancer, et impose une surveillance plus rapprochée.
• L’inflammation aiguë peut être responsable d’une colite aiguë grave dans les deux maladies, plus fréquemment dans la RCH.
• Dans la MC, l’inflammation aiguë transmurale peut entraîner l’apparition d’abcès, une péritonite ou une fistule dans un autre organe : anse grêle, sigmoïde, vessie, voire la peau (chez les malades déjà opérés).
•
Points clés
• La maladie de Crohn et la rectocolite hémorragique sont des maladies inflammatoires chroniques de l’intestin qui évoluent par poussées.
• Le diagnostic repose sur un faisceau d’arguments cliniques, biologiques, endoscopiques, anatomopa-thologiques et radiologiques.
• Leur diagnostic nécessite la réalisation de biopsies étagées avec examen anatomopathologique lors d’une iléocoloscopie.
• Les biopsies doivent être multiples, étagées, en muqueuse d’aspect pathologique et sain.
• Le risque de dysplasie augmente avec la durée de la maladie, son étendue et la persistance d’une inflammation active.
• Après le diagnostic initial, la réalisation d’une nouvelle endoscopie avec biopsies étagées est justifiée pour rechercher des complications :
– lors d’une poussée sévère ou non contrôlée par le traitement, pour juger de l’étendue et de la sévérité des lésions, et rechercher une infection par le CMV associée ;
– lors du suivi de la maladie, à la recherche de lésions précancéreuse de dysplasie épithéliale ou d’un adénocarcinome.
Item 285 – Diarrhée chronique chez l’adulte et l’enfant
Auteure : Camille Boulagnon-Rombi Camille
I. Prérequis
II. Diarrhée chronique : définition et principales étiologies
III. Maladie cœliaque
IV. Colite microscopique
Hiérarchisation des connaissances Tableau 5
{kind=link}
I. Prérequis
Histologie de l’intestin grêle et du côlon (cf. supra item 282 – Maladies inflammatoires chroniques de l’intestin chez l’adulte).
II. Diarrhée chronique : définition et principales étiologies
En cas de doute diagnostique, une diarrhée est définie par un poids moyen des selles supérieur à 300 g/24 h (pesée faite sur 3 jours consécutifs).
La diarrhée chronique est définie par une durée supérieure à 4 semaines.
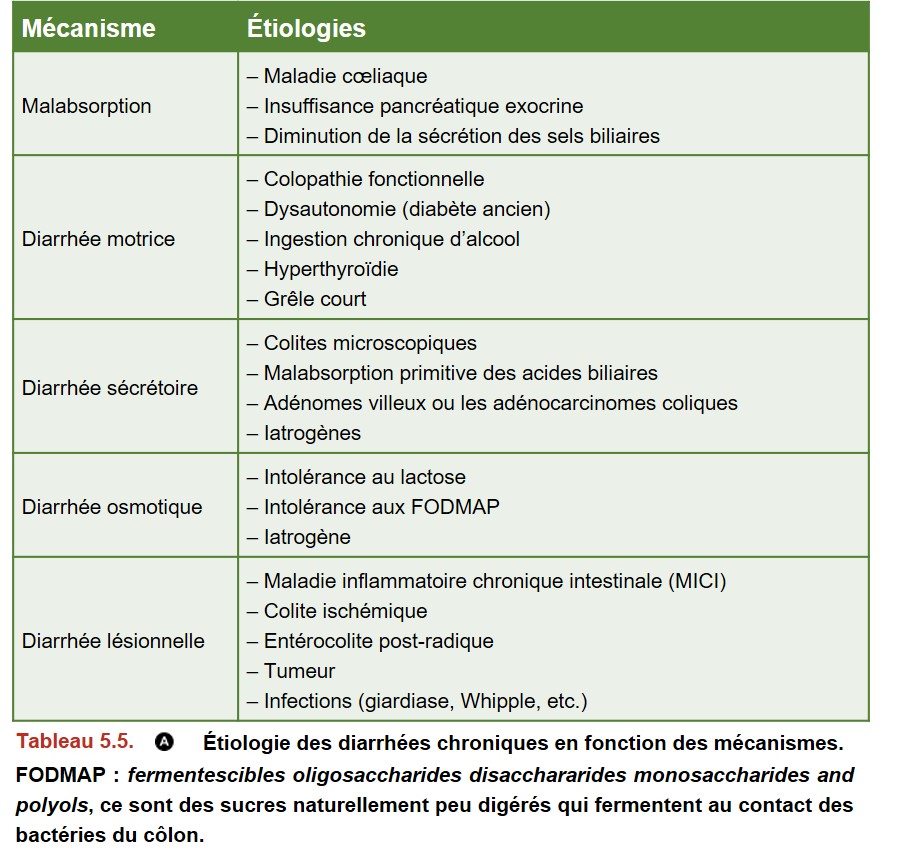
Différents mécanismes physiopathologiques sont impliqués dans les diarrhées chroniques :
• la diarrhée sécrétoire ;
• la diarrhée motrice ;
• la diarrhée osmotique ;
• la diarrhée par malabsorption ;
• la diarrhée lésionnelle.
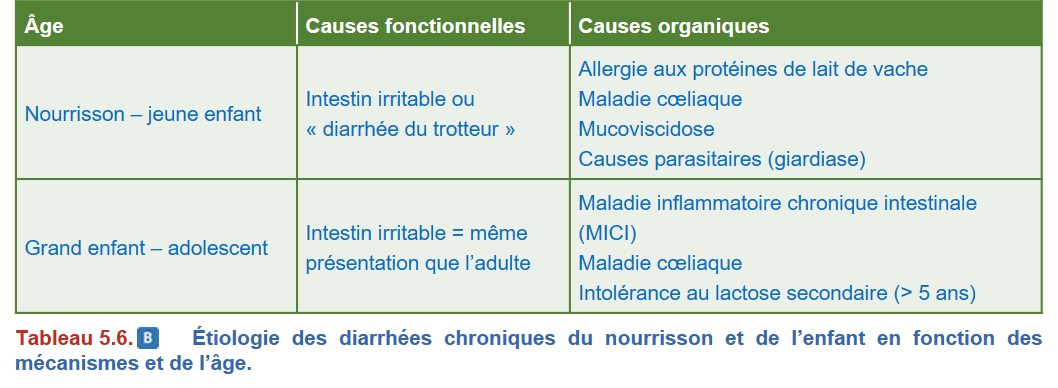
Le mécanisme impliqué, l’âge du patient et ses antécédents permettent d’envisager certaines étiologies (Tableau 5.5 et Tableau 5.6).
{kind=link}
{kind=link}
L’exploration endoscopique digestive avec EOGD et iléocoloscopie est indiquée :
• en cas de diarrhée d’allure lésionnelle ;
• en l’absence de cause évidente ;
• en cas de signes d’alarme (rectorragie, altération de l’état général, modification récente et inexpliquée du transit) ;
• si l’âge est > 50 ans.
Des biopsies systématiques doivent être faites au niveau :
• du duodénum : recherche d’atrophie (maladie cœliaque) ;
• de l’estomac : recherche de gastrite chronique atrophique (maladie de Biermer) ;
• du côlon : recherche d’une colite microscopique en cas d’aspect endoscopique normal ;
• de l’iléon terminal : recherche d’arguments en faveur d’une MICI (Crohn).
III. Maladie cœliaque
A. Épidémiologie et terrain
B. Diagnostic
Les 4 lésions anatomopathologiques clés au niveau de la muqueuse duodénale (Figure 5.13)
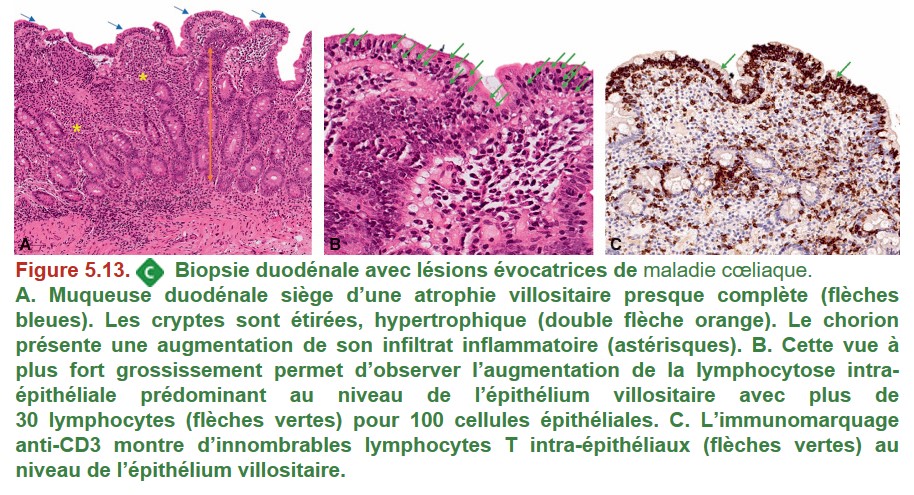{kind=link}
• Augmentation des lymphocytes intra-épithéliaux
• Atrophie villositaire totale ou subtotale
• Hypertrophie des cryptes
• Infiltrat lymphocytaire et plasmocytaire du chorion augmenté
D’autres causes de diarrhée (giardiose, strongyloïdose, maladie de Whipple, infections associées à un déficit immunitaire [cryptococcose, isosporose]) peuvent également être diagnostiquée par l’histologie. L’histologie est nécessaire au diagnostic de maladie cœliaque mais ne suffit pas. En effet, d’autres pathologies peuvent donner des lésions histologiques comparables avec atrophie villositaire et/ou augmentation des lymphocytes intra-épithéliaux (sprue tropicale, déficits immunitaires, médicaments comme l’olmésartan, etc.).
C. Complications
• la sprue réfractaire (entité prélymphomateuse) ;
• le lymphome T du grêle ;
• certains cancers épithéliaux (œsophage, pharynx, grêle).
• en premier lieu, une mauvaise observance du régime sans gluten (nouvelles biopsies duodénales pour analyse histologique, nouveau dosage des IgA anti-transglutaminase, interrogatoire par diététicien) ;
• une remise en cause du diagnostic de maladie cœliaque (diagnostic différentiel d’une atrophie villositaire) ou l’association à une autre maladie comme une colite microscopique pouvant être associée à la maladie cœliaque et qui peut expliquer une diarrhée persistante ou récidivante (nécessité de biopsies avec examen anatomopathologique pour le diagnostic de colite microscopique) ;
• une néoplasie du grêle (lymphome, adénocarcinome, sprue réfractaire) sur biopsie (diagnostic anatomopathologique).
IV. Colite microscopique
Les colites microscopiques sont responsables d’une diarrhée sécrétoire, aqueuse. Cette maladie reste rare (fréquence proche de celle des MICI) et est souvent d’origine iatrogène. Elle peut être déclenchée par des médicaments inhibiteurs de la pompe à protons (lansoprazole), les AINS, les inhibiteurs de la recapture de la sérotonine, les veinotoniques principalement ou être associées à d’autres maladies auto-immunes (polyarthrite rhumatoïde, thyroïdite, syndrome de Sjögren, etc.), dont la maladie cœliaque ou la gastrite auto-immune.
La présentation associe :
• une diarrhée aqueuse (de début brutal dans 50 % des cas) ;
• un aspect endoscopique normal de la muqueuse colique (d’où le terme de colite « microscopique » car elle ne se voit qu’au microscope).
Le diagnostic de colite microscopique nécessite la réalisation de biopsies étagées multiples au cours d’une coloscopie totale.
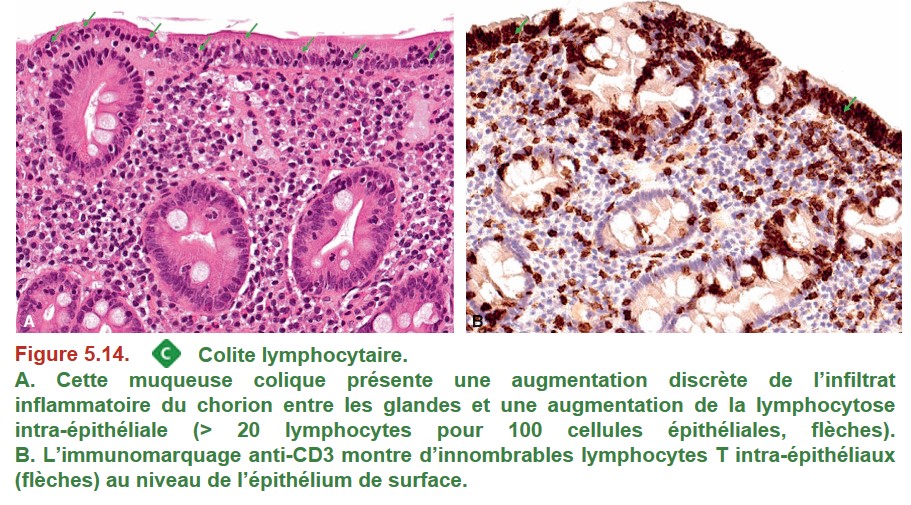{kind=link}
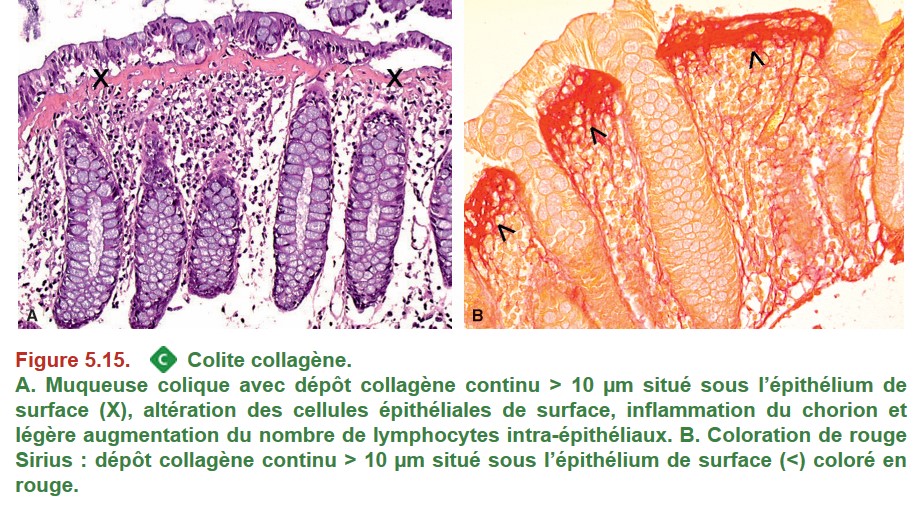{kind=link}
• la colite collagène : augmentation de l’épaisseur de la bande collagène sous-épithéliale ;
• la colite lymphocytaire : augmentation du nombre des lymphocytes intra-épithéliaux.
Points clés
Diarrhée chronique
• L’exploration endoscopique par EOGD et iléocoloscopie avec réalisation de biopsies est indiquée :
– en cas de diarrhée d’allure lésionnelle ;
– en l’absence de cause évidente ;
– en cas de signes d’alarme (rectorragie, altération de l’état général, modification récente et inexpli-quée du transit) ;
– si l’âge est > 50 ans.
• Des biopsies systématiques sont faites au niveau du duodénum, de l’estomac, du côlon et de l’iléon terminal.
Maladie cœliaque
• Le diagnostic repose sur l’association de critères anatomopathologiques et sérologiques.
• L’atrophie villositaire avec augmentation du nombre de lymphocytes intra-épithéliaux, une hypertrophie des cryptes avec un infiltrat lymphocytaire et plasmocytaire du chorion augmenté sont évocateurs au niveau histologique.
• La sprue réfractaire, le lymphome ou les cancers épithéliaux peuvent compliquer la maladie cœliaque.
Colite microscopique
• L’iléocoloscopie ne révèle aucune lésion macroscopique.
• Le diagnostic est histologique et nécessite des biopsies coliques multiples et étagées.
• Deux entités sont à distinguer :
– colite lymphocytaire avec augmentation du nombre de lymphocytes intra-épithéliaux ;
– colite collagène avec augmentation de l’épaisseur de la bande collagène sous-épithéliale.
Item 305 – Tumeurs de l’œsophage
Auteure : Charlotte Dufour
I. Prérequis
II. Types histologiques, lésions précancéreuses
III. Diagnostic
IV. Anatomie pathologique et exérèse à but thérapeutique
Hiérarchisation des connaissances Tableau 6
{kind=link}
I. Prérequis
Il faut connaître l’histologie de la paroi œsophagienne (cf. Figure 5.1).
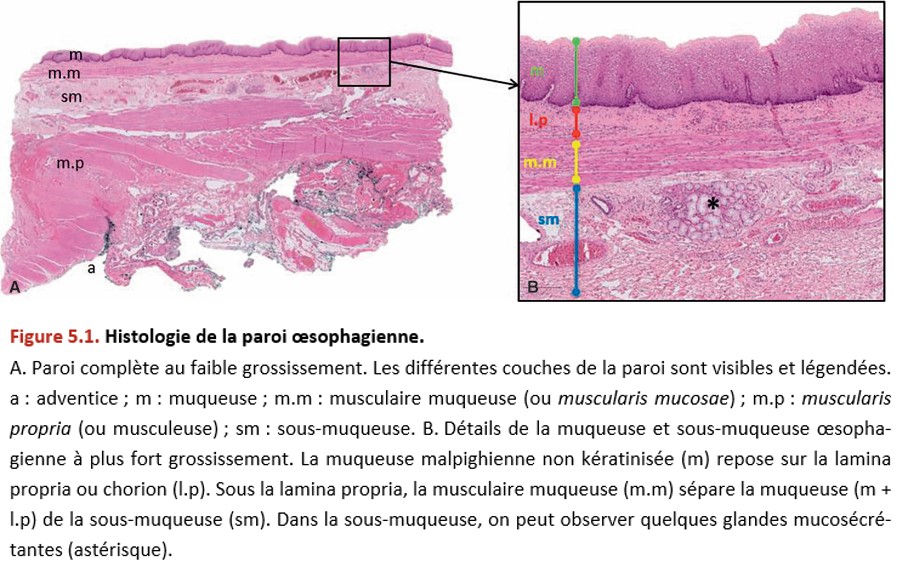{kind=link}
II. Types histologiques, lésions précancéreuses
A. Carcinome épidermoïde
Le carcinome épidermoïde est une tumeur épithéliale maligne à différenciation malpighienne. Il correspond à une transformation maligne de l’épithélium malpighien tapissant normalement la paroi œsophagienne (Figure 5.16).
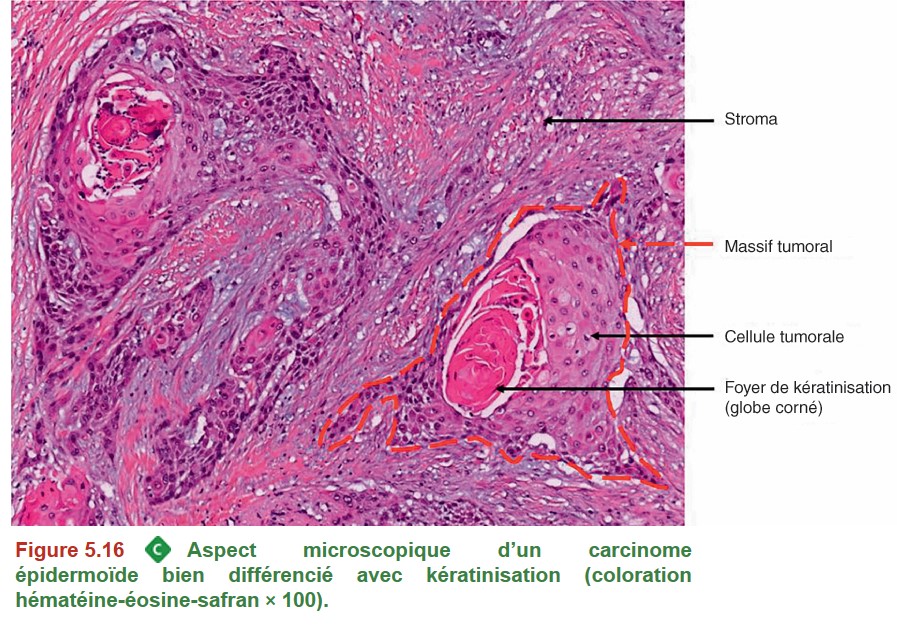{kind=link}
Les deux principaux facteurs de risque de survenue sont le tabac et l’alcool (également impliqués dans les cancers de la sphère ORL et bronchopulmonaires). Certaines conditions pathologiques, telles que l’achalasie (trouble moteur œsophagien, responsable d’une dysphagie), l’œsophagite caustique ou post-radique exposent également au risque de survenue de carcinome épidermoïde. L’ingestion de boissons brûlantes a aussi été mise en cause. On constate par ailleurs une diminution de l’incidence du carcinome épidermoïde depuis plusieurs décennies, en rapport avec l’évolution de l’exposition aux facteurs de risque.Le carcinome épidermoïde peut se développer sur toute la hauteur de l’œsophage.
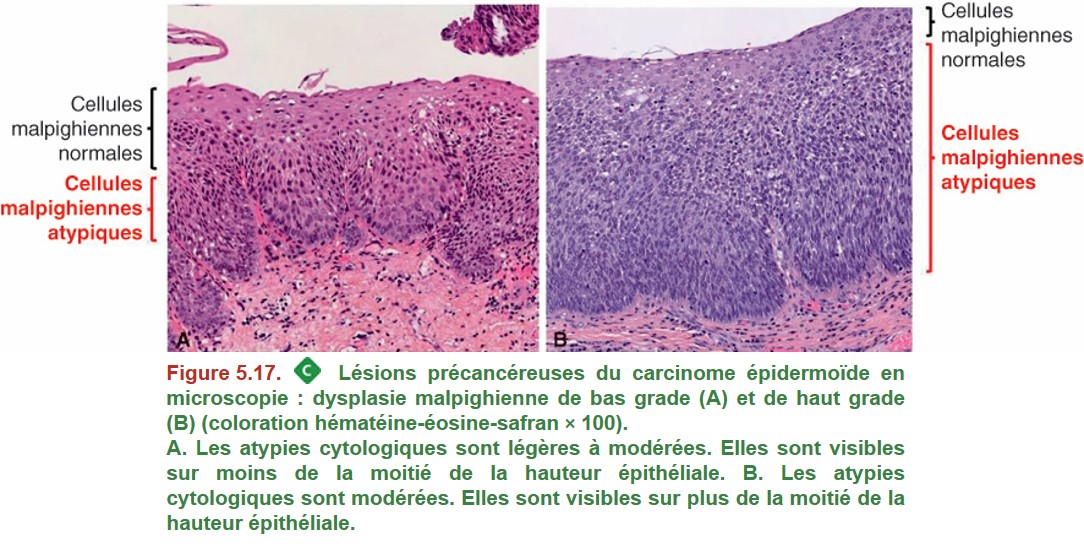{kind=link}
La dysplasie épithéliale malpighienne reste cantonnée au sein de l’épithélium de surface, sans franchissement de la membrane basale. Une fois celle-ci franchie, il s’agit alors d’un carcinome épidermoïde infiltrant. Le degré de dysplasie est défini en fonction des atypies cellulaires et de la désorganisation architecturale de l’épithélium.
B. Adénocarcinome de l’œsophage
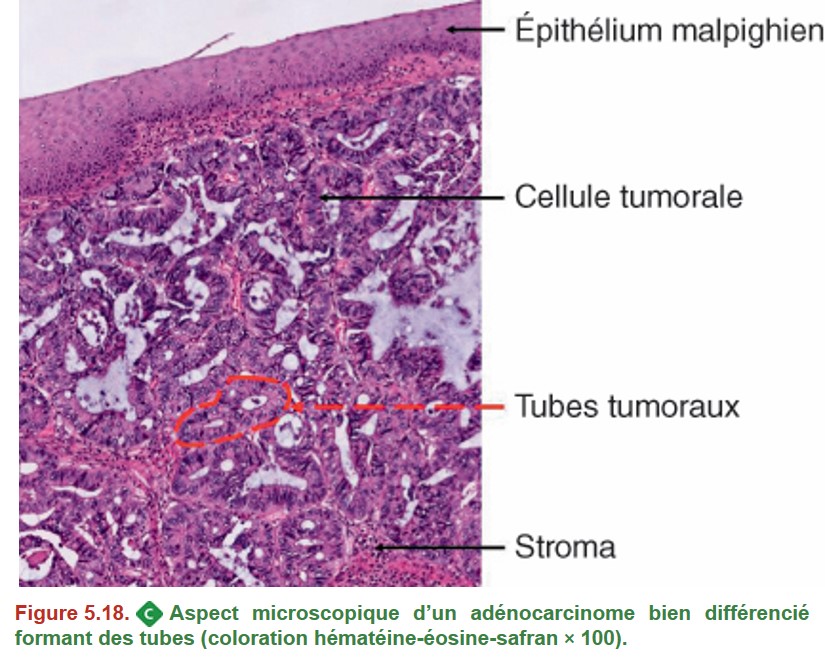{kind=link}
Il est le plus souvent situé au niveau du bas œsophage ou de la jonction œsogastrique.
III. Diagnostic
IV. Anatomie pathologique et exérèse à but thérapeutique
Au diagnostic, le bilan d’extension ainsi que l’évaluation de l’état général du patient permettent d’orienter la stratégie thérapeutique. Celle-ci est définie en réunion de concertation pluridisciplinaire.
A. Pièce de mucosectomie ou de dissection sous-muqueuse endoscopique à but curatif
Le traitement endoscopique à visée curative est réservé aux tumeurs superficielles (envahissement au maximum de la sous-muqueuse), a priori sans envahissement ganglionnaire ni métastase au diagnostic. Deux techniques sont possibles : la mucosectomie ou la dissection sous-muqueuse. Le choix de la technique dépend du type histologique de la tumeur (carcinome épidermoïde ou adénocarcinome) et de sa taille. Les mucosectomies ou dissections sous-muqueuses endoscopiques se font par :
• repérage et délimitation de la lésion ;
• injection d’un liquide dans la sous-muqueuse, permettant de soulever la muqueuse ;
• résection de la lésion avec une marge de muqueuse saine.
Le prélèvement intéresse donc la muqueuse et la sous-muqueuse. Idéalement, la pièce de résection doit être adressée au laboratoire, épinglée sur un support et orientée, afin d’optimiser son analyse anatomopathologique.
L’examen anatomopathologique précise :
• le type histologique selon la classification de l’OMS en vigueur, avec le grade de différenciation de la tumeur ;
• la présence d’emboles tumoraux ou d’engainements périnerveux tumoraux (identifiés/non identifiés) ;
• le niveau d’infiltration de la tumeur dans la paroi avec la mesure de la hauteur d’infiltration, notamment en cas d’atteinte de la sous-muqueuse ;
• la qualité de l’exérèse (limite saine : oui/non. Si oui, mesure des marges profondes et latérales [en mm]).
En fonction des résultats de l’examen anatomopathologique, des traitements complémentaires ou une surveillance simple sont proposés.
B. Pièce de chirurgie d’exérèse à but curatif
Elle n’est réalisée que chez des patients opérables avec une lésion résécable, d’emblée ou non. En cas de stade précoce (atteinte au maximum de la musculeuse, sans envahissement ganglionnaire), la chirurgie seule est proposée. En cas de stade localement avancé (dépassement de la musculeuse et/ou envahissement ganglionnaire), une radiochimiothérapie néoadjuvante ou une chimiothérapie périopératoire (adénocarcinome) sont indiquées. L’intervention de choix pour les tumeurs du tiers moyen et du tiers inférieur de l’œsophage est l’œsophagectomie transthoracique subtotale avec curage ganglionnaire et plastie gastrique (Figure 5.19). Cette opération est réalisée selon la technique de Lewis Santy. La pièce opératoire est adressée au laboratoire d’anatomie pathologique.
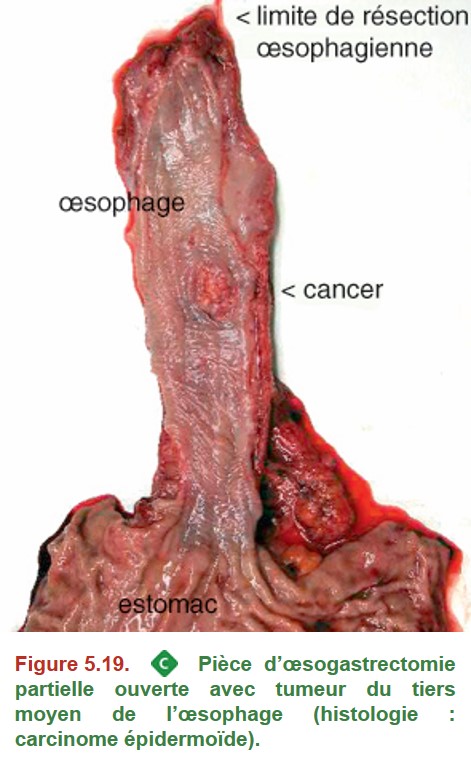{kind=link}
L’examen anatomopathologique doit préciser (données minimales définies avec l’INCa, 2011) :
• le type de pièce opératoire, la localisation de la tumeur ;
• le type histologique de la tumeur (suivant la classification de l’OMS en vigueur) ;
• le grade de différenciation ;
• la réponse au traitement néoadjuvant éventuel ;
• le niveau d’infiltration de la tumeur dans la paroi ;
• l’extension tumorale dans les ganglions régionaux (nombre de ganglions envahis/nombre de ganglions prélevés) ;
• la présence ou non d’emboles vasculaires ou d’engainements périnerveux ;
• la qualité de l’exérèse avec les marges (limites proximales et distales/limite circonférentielle) ;
• le stade pTNM en précisant l’année de la classification utilisée.
Points clés
• Le diagnostic de tumeur de l’œsophage nécessite des biopsies multiples avec examen anatomopathologique qui précise le type histologique.
• Les deux types histologiques les plus fréquents sont l’adénocarcinome et le carcinome épidermoïde.
• Le carcinome épidermoïde infiltrant est précédé d’une lésion dysplasique.
• L’adénocarcinome du bas œsophage se développe souvent sur des lésions d’endobrachyœsophage (séquence métaplasie – dysplasie – carcinome).
• En cas d’adénocarcinome, une recherche d’instabilité des microsatellites et le statut HER2 doivent être évalués.
Item 301 – Tumeurs du côlon et du rectum
Auteurs : Florence Renaud, Dominique Wendum
I. Prérequis : histologie de la paroi colique et rectale
II. Adénocarcinome colorectal
Hiérarchisation des connaissances Tableau 7
{kind=link}
I. Prérequis : histologie de la paroi colique et rectale
Cf. supra item 282 – Maladies inflammatoires chroniques de l’intestin (MICI) chez l’adulte.
II. Adénocarcinome colorectal
A. Épidémiologie
{kind=link}
Les modalités de dépistage sont différentes dans ces trois groupes :
• dépistage de masse pour les sujets sans facteur de risque (risque moyen) par test immunologique de recherche de sang dans les selles (FIT : fecal immunologic test) tous les 2 ans à partir de l’âge de 50 ans et jusqu’à 75 ans ;
• coloscopies itératives chez sujets à risque élevé et très élevé (en cas d’antécédent personnel ou familial de CCR ou d’adénome, de MICI ancienne, de prédisposition héréditaire, de syndrome de Lynch ou de polypose familiale, d’acromégalie).
B. Types histologiques
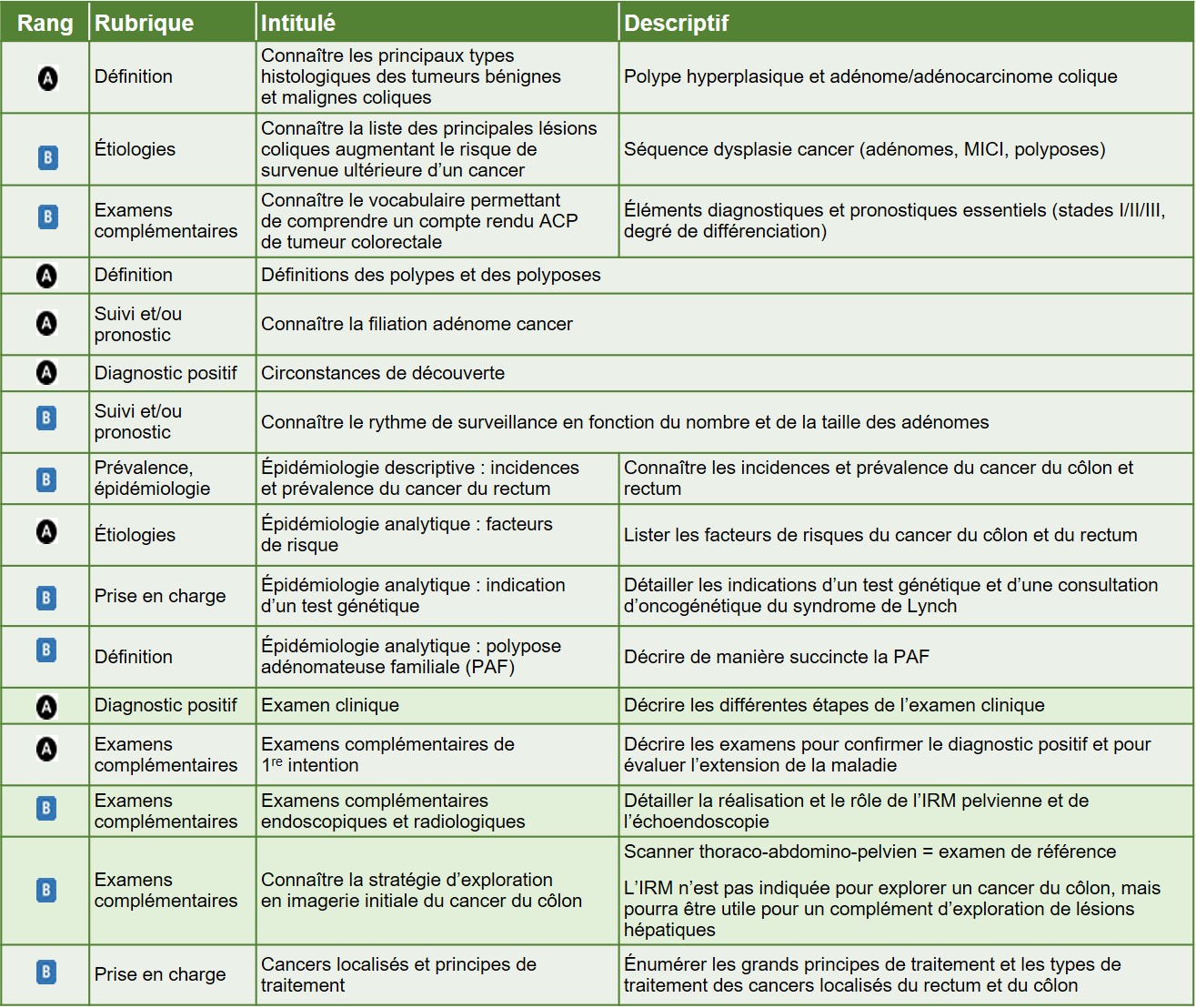
Plus de 95 % des CCR sont des adénocarcinomes. Le plus fréquent est l’adénocarcinome lieberkühnien +++ (Figure 5.20). Il est classé en fonction de sa différenciation (c’est-à-dire de sa ressemblance avec le tissu normal, évaluée ici en fonction du pourcentage de structures glandulaires dans la tumeur) et peut être :
{kind=link}
• de bas grade (bien ou moyennement différencié) ;
• de haut grade (peu différencié).
Le pronostic est moins bon avec la perte de différenciation.
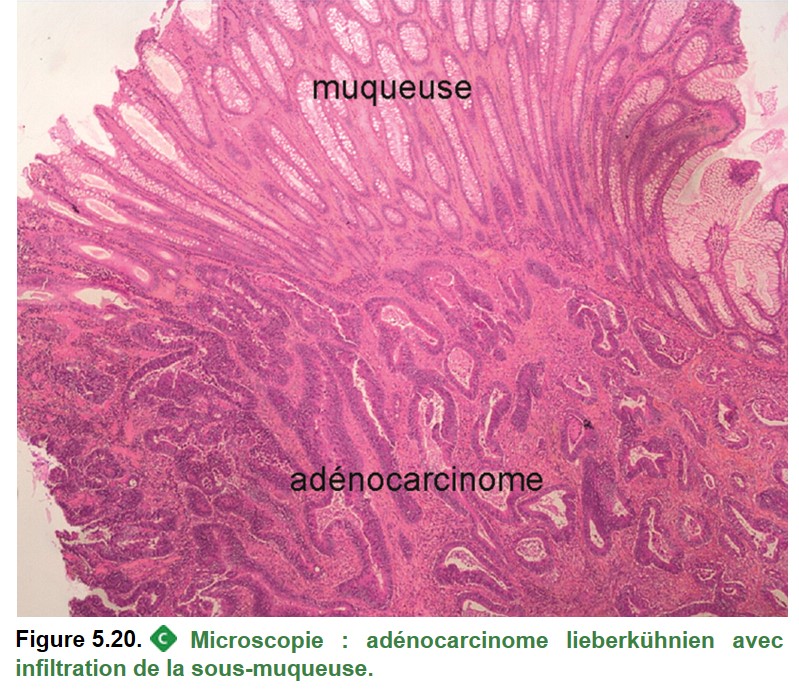
• d’un adénocarcinome colloïde (mucineux) : larges flaques de mucus dans plus de 50 % de la tumeur (Figure 5.21) ;
{kind=link}
• d’un carcinome médullaire : cellules très cohésives à limites cytoplasmiques imprécises et sans structure glandulaire ;
Remarque : Ce sous-type n’a aucun rapport avec le carcinome médullaire de la thyroïde malgré la même dénomination. Ce type de cancer présente en général une instabilité des microsatellites (cf. infra).
• d’un adénocarcinome à cellules en bague à chaton, etc.
C. Lésions précancéreuses, cancérogenèse
Rappels
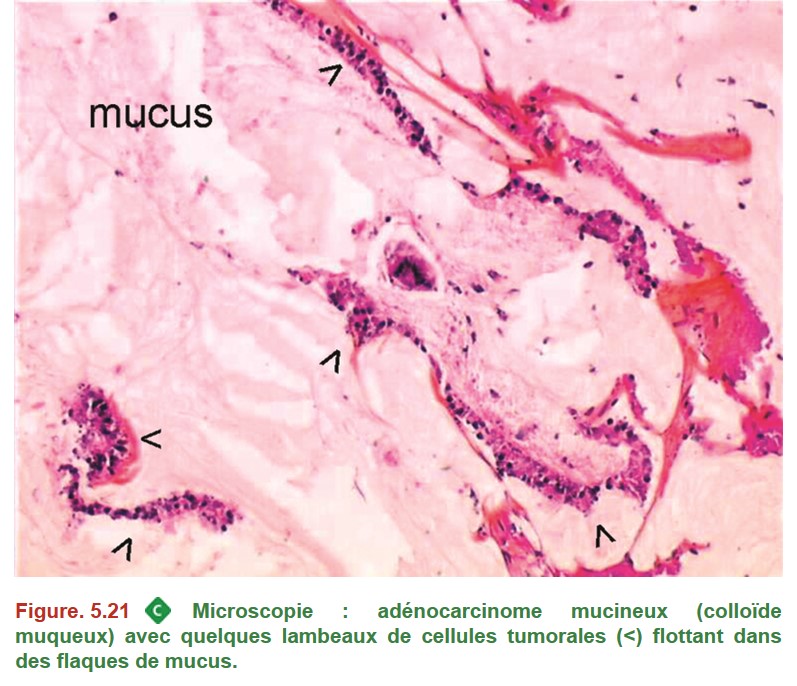
• Un polype est défini en macroscopie comme une lésion qui fait saillie dans la lumière du tube digestif. Le caractère dysplasique ou non est déterminé par l’examen microscopique (dans le tube digestif, polype dysplasique = adénome).
• Une polypose est caractérisée par la présence de nombreux polypes. Si le seuil de plus de 100 polypes adénomateux est classiquement défini pour la polypose adénomateuse familiale, il n’est pas possible de donner un nombre minimal de polypes qui permettrait de définir une polypose de manière générale car cela dépend du contexte (type de polype, âge du patient, taille et localisation des polypes, etc.).
1. Principaux syndromes de prédisposition génétique
Polypose adénomateuse familiale (PAF)
{kind=link}
• 1 % des CCR, mutation germinale du gène APC (transmission autosomique dominante, péné-trance complète).
• Développement de multiples polypes adénomateux colorectaux (> 100 polypes) avec un risque de 100 % de développer un CCR (cancer avec instabilité chromosomique). Il est important de noter qu’environ 20 % des mutations du gène APC surviennent de novo (sans histoire familiale connue), résultant d’une mutation acquise lors de la gamétogenèse chez l’un des parents.
Les autres manifestations tumorales sont :
• les adénomes duodénaux surtout périampullaires pouvant se transformer en cancer ;
• les polypes glandulokystiques gastriques (cf. infra item 303 – Tumeurs de l’estomac) ;
• la tumeur desmoïde : prolifération tumorale de myofibroblastes (le plus souvent dans la paroi abdominale ou dans le mésentère). Cette tumeur est très infiltrante mais ne donne pas de métastase. C’est une tumeur dite à malignité locale qui peut entraîner le décès.
Polypose associée à MUTYH (MAP)
• Mutation biallélique du gène MUTYH (transmission autosomique récessive, pénétrance quasi complète), gène de réparation de l’ADN appartenant au système de réparation BER (base exci-sion repair), impliqué dans le processus de réparation des lésions oxydatives de l’ADN.
• Polypes multiples du côlon (souvent polypose atténuée, entre 5 et 100 polypes), mais aussi du duodénum et de l’estomac.
• Risque cumulé de cancer de l’ordre de 10 à 50 %.
Syndrome de Lynch
• Deux à 5 % des CCR, mutation d’un des gènes MMR (le plus souvent MLH1, MSH2, plus rare-ment PMS2 ou MSH6), entraînant une instabilité de microsatellites (cf. infra).
• Risque très élevé de développer un CCR (40 à 70 % de risque cumulé de CCR pour les hommes et 20 à 50 % pour les femmes), âge moyen de survenue 45-50 ans.
• Histologiquement, les adénocarcinomes liés à une mutation d’un gène MMR présentent souvent des caractéristiques morphologiques particulières (mucineux ou médullaires).
• Autres localisations tumorales : endomètre, voies urinaires, intestin grêle, estomac, ovaires, cholangiocarcinome.
Polyposes hamartomateuses
Très rares, elles sont caractérisées par la présence de polypes hamartomateux du tractus gastro-intestinal avec un risque de développer un CCR. Ces syndromes sont parfois associés au développement d’autres cancers.
Syndrome de Peutz-Jeghers
• Mutation du gène LKB1/STK11.
• Lésions cutanées pigmentées typiques (lentiginose périorificielle).
• Risque de cancer associé : ovaire, pancréas, grêle, etc.
Polypose juvénile familiale
• Mutation du gène SMAD4 ou du gène BMPR1A.
• Multiples polypes hamartomateux du côlon, du rectum, de l’estomac et de l’intestin grêle avec risque de développer un cancer dans ces organes.
Maladie de Cowden (ou syndrome des hamartomes multiples)
• Mutation du gène PTEN.
• Polypes hamartomateux gastro-intestinaux, mais aussi de la peau, de la thyroïde.
• Risque de développer un cancer colique, également de la thyroïde, du sein et de l’endomètre.
2. Deux principales voies de cancérogenèse
Il existe principalement deux types de cancers colorectaux en fonction de leurs anomalies génétiques :
• les cancers avec instabilité chromosomique (environ 85 % des cancers) ;
• les cancers avec instabilité des microsatellites (15 % des cancers, appelés aussi cancers MSI+).
Les cancers avec instabilité des microsatellites sont des cancers liés à une déficience du système de réparation des mésappariements (MMR) qui contrôle la fidélité de la réplication de l’ADN. La conséquence directe de ce défaut fonctionnel est l’accumulation de mutations dans les cellules tumorales, en particulier au niveau de séquences répétées de 1 à 5 nucléotides appelées séquences microsatellites. Deux protéines du système de réparation de l’ADN sont très fréquemment impliquées dans la genèse des tumeurs MSI+ : hMLH1, hMSH2. Il y a dans les cellules tumorales une perte d’expression protéique de hMLH1 ou de hMSH2 responsable de l’instabilité génétique au niveau des séquences microsatellites. Les cancers MSI+ peuvent être sporadiques (10 à 15 % des cancers) ou familiaux (2 à 5 % des cancers [syndrome de Lynch]). Les cancers développés sur PAF sont des cancers avec instabilité chromosomique.
3. Principale lésion tissulaire précancéreuse : l’adénome
{kind=link}
L’adénome est une tumeur épithéliale glandulaire bénigne.
On distingue les adénomes « sans autre précision » (Figure 5.24) des lésions festonnées (« lésions festonnées sessiles » ou « adénomes festonnés traditionnels suivant la terminologie actuelle) (Figure 5.25).
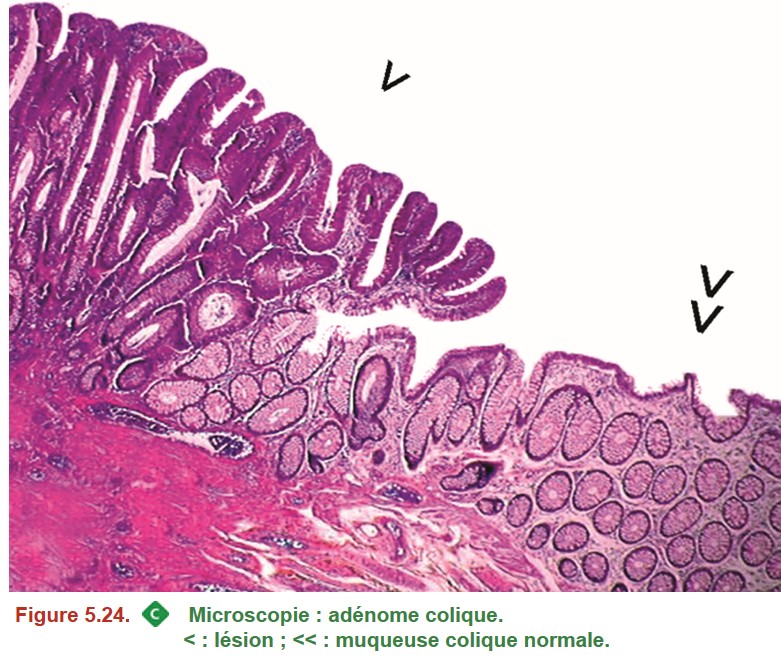{kind=link}
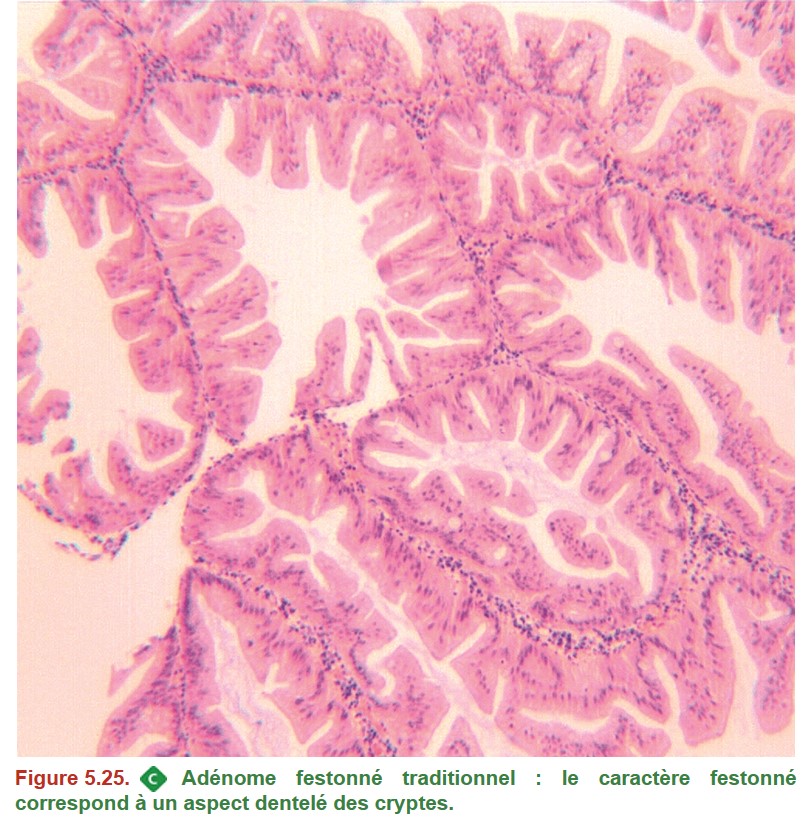{kind=link}
Les adénomes sont classés en fonction de leur :
• aspect endoscopique (sessiles, pédiculés, plans) ;
• architecture microscopique (tubuleux : > 80 % de tubes ; tubulovilleux, villeux : > 80 % de vil-losités) ; certains adénomes ont une architecture festonnée (dentelée), ils sont classés à part ;
• degré de dysplasie (bas grade, haut grade, en fonction du degré d’anomalies cytonucléaires et architecturales).
Tous les adénomes ne cancérisent pas inéluctablement. On estime que seuls 10 % des adénomes atteignent 1 cm ou plus. Globalement, seulement un quart des adénomes de plus de 1 cm deviennent des cancers.
La transformation d’un adénome en adénocarcinome se fait dans un délai estimé de 10 à 20 ans, délai qui peut être plus court en cas d’adénome ou lésion sessile festonnée (MSI+).
Le risque de transformation des adénomes en cancer est augmenté en fonction :
• du degré de dysplasie (haut grade) ;
• de la taille du polype adénomateux (> 1 cm), risque quasi nul de cancer sur un polype adénomateux < 1 cm ;
• de l’architecture (villeuse ou adénome plan).
À l’endoscopie, tout polype doit être réséqué (lorsque c’est possible), et envoyé en anatomopathologie.
L’examen anatomopathologique précise :
• le type histologique du polype (adénome, adénome festonné, polype hyperplasique, hamartome, etc.) ;
• en cas d’adénome : l’architecture, le degré de dysplasie, la présence ou non d’un foyer d’adénocarcinome associé.
Si un polype adénomateux présente un foyer d’adénocarcinome invasif (dépassant la musculaire muqueuse, c’est-à-dire atteignant au moins la sous-muqueuse), on parle de polype cancérisé ou transformé.
Attention ! À la différence des autres organes, au niveau colorectal, un cancer infiltrant uniquement la muqueuse n’est pas dit infiltrant mais in situ (alors que la membrane basale est franchie). C’est parce qu’au niveau du côlon et du rectum, contrairement aux autres organes, le risque de dissémination d’un cancer strictement intramuqueux est nul : il est donc assimilé à un cancer in situ dans la classification TNM (Tis).
Les polypes adénomateux peuvent se cancériser. Leur exérèse est donc un traitement préventif du cancer. Si la lésion vue en endoscopie a de fortes chances de comporter un foyer de cancer (lésion > 1 cm, architecture villeuse ou plane), l’exérèse du polype doit permettre au pathologiste de préciser les critères pronostiques de cet éventuel cancer pour pouvoir décider ensuite de la réalisation d’une éventuelle colectomie complémentaire.
Ces critères pronostiques sont :
• le type histologique de cancer, le degré de différenciation ;
• la présence ou non d’emboles tumoraux vasculolymphatiques ;
• le degré de bourgeonnement tumoral au niveau du front d’invasion (budding = cellules tumorales isolées ou en petits amas) ;
• la profondeur d’invasion du cancer dans la sous-muqueuse ;
• la qualité de l’exérèse avec mesure des marges.
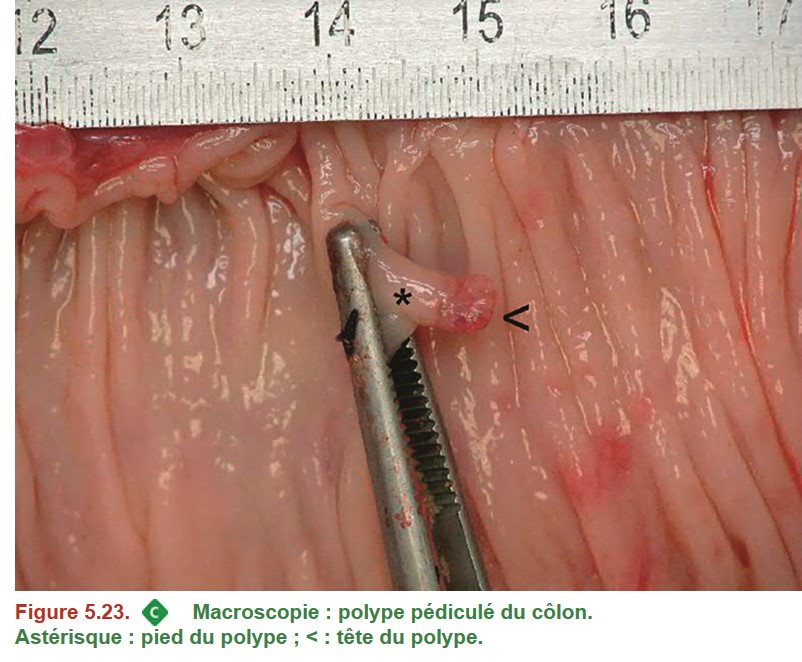
En cas de polype pédiculé, il faut donc que le pied du polype soit repéré.
En cas de mucosectomie, celle-ci doit être épinglée sur un support (Figure 5.26).
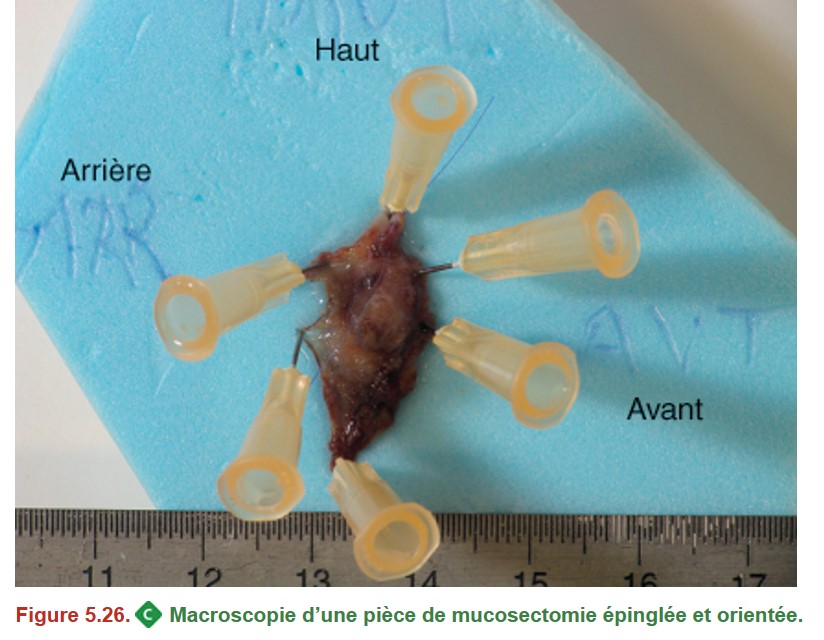{kind=link}
4. Autres types de polypes
Les principaux types de polypes colorectaux non adénomateux sont :
• les polypes hyperplasiques ;
• les polypes hamartomateux (rares) ;
• les « pseudo-polypes » au cours des colites.
Remarque : Les polypes dits hyperplasiques prédominent dans le côlon distal et le rectum. Ils font partie des polypes festonnés mais ils n’ont pas de risque de dégénérescence, contrairement aux lésions sessiles festonnés et aux adénomes festonnés traditionnels.
Les polypes hamartomateux sont rares (polypes juvéniles, polypes de type Peutz-Jeghers) et peuvent se voir de manière sporadique en dehors de toute polypose hamartomateuse. Il existe un risque de dégénérescence.Les « pseudo-polypes » (cf. infra) sont des polypes constitués d’une zone de muqueuse en régénération, entre deux zones ulcérées, réalisant donc une saillie dans la lumière colique.
D. Diagnostic
Le compte rendu anatomopathologique affirme le diagnostic de cancer et précise le type histologique.
En effet, toutes les tumeurs colorectales ne sont pas obligatoirement des adénocarcinomes. Bien que cela soit rare, il peut s’agir d’autres types histologiques dont le traitement diffère de celui d’un adénocarcinome (ex : lymphome, sarcome, tumeur neuroendocrine).
E. Principes du traitement et anatomopathologie
1. Exérèse
En cas de tumeur résécable, le traitement à visée curative des adénocarcinomes du côlon et du haut rectum repose sur l’exérèse chirurgicale carcinologique, permettant des marges de 5 cm de part et d’autre de la tumeur, ainsi que l’exérèse du mésocôlon contenant les ganglions lymphatiques (12 ganglions minimum) (Figure 5.27). Pour les tumeurs du rectum, le choix entre intervention conservatrice (proctectomie avec anastomose colorectale ou colo-anale) ou amputation du rectum et de l’anus (amputation abdomino-périnéale avec colostomie définitive) dépend du siège de la tumeur. Une radiochimiothérapie préopératoire (traitement néoadjuvant) est indiquée pour les cancers du moyen et bas rectum localement avancés (tumeurs T3 ou T4 et/ou N+).
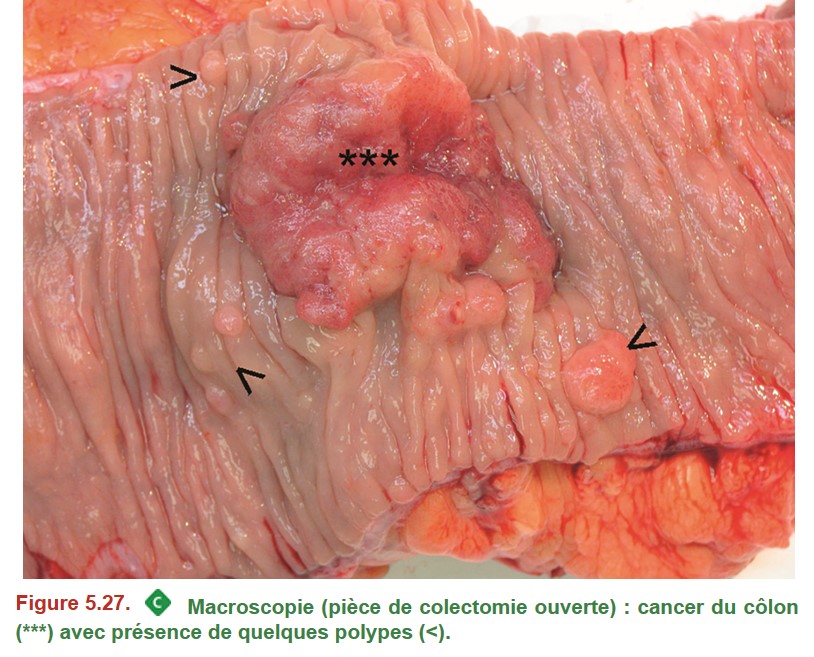{kind=link}
2. Examen anatomopathologique de la pièce opératoire
Il contribue également à la prise en charge en évaluant le pronostic et en définissant des critères importants pour la prescription d’un éventuel traitement complémentaire postopératoire. Une chimiothérapie adjuvante est proposée en cas cancer du côlon stade III (présence de métastase ganglionnaire identifiée à l’analyse anatomopathologique). Le curage doit avoir intéressé au moins 12 ganglions.
• le type histologique de la tumeur (suivant la classification de l’OMS en vigueur) avec son grade de différenciation ;
• les éléments permettant de donner le pTNM de la tumeur :
– critères relatifs à la tumeur :
– degré d’infiltration de la paroi colorectale (ou du mésorectum) et des organes adjacents,
– présence d’une perforation en zone tumorale,
– présence de dépôts tumoraux dans le méso,
– critères relatifs aux ganglions : nombre de ganglions envahis/nombre de ganglions prélevés,
– présence ou non d’engainements périnerveux ou d’emboles vasculaires,
– qualité de l’exérèse (limites proximales et distales saines : oui/non),
– pour les cancers du rectum : marge circonférentielle, c’est-à-dire la distance entre la tumeur et la limite d’exérèse chirurgicale latérale dans le mésorectum permettant d’évaluer la qualité de l’exérèse (R0 ou R1),
– en cas de traitement néoadjuvant : réponse au traitement (régression tumorale).
{kind=link}
3. Recherche d’une instabilité des microsatellites
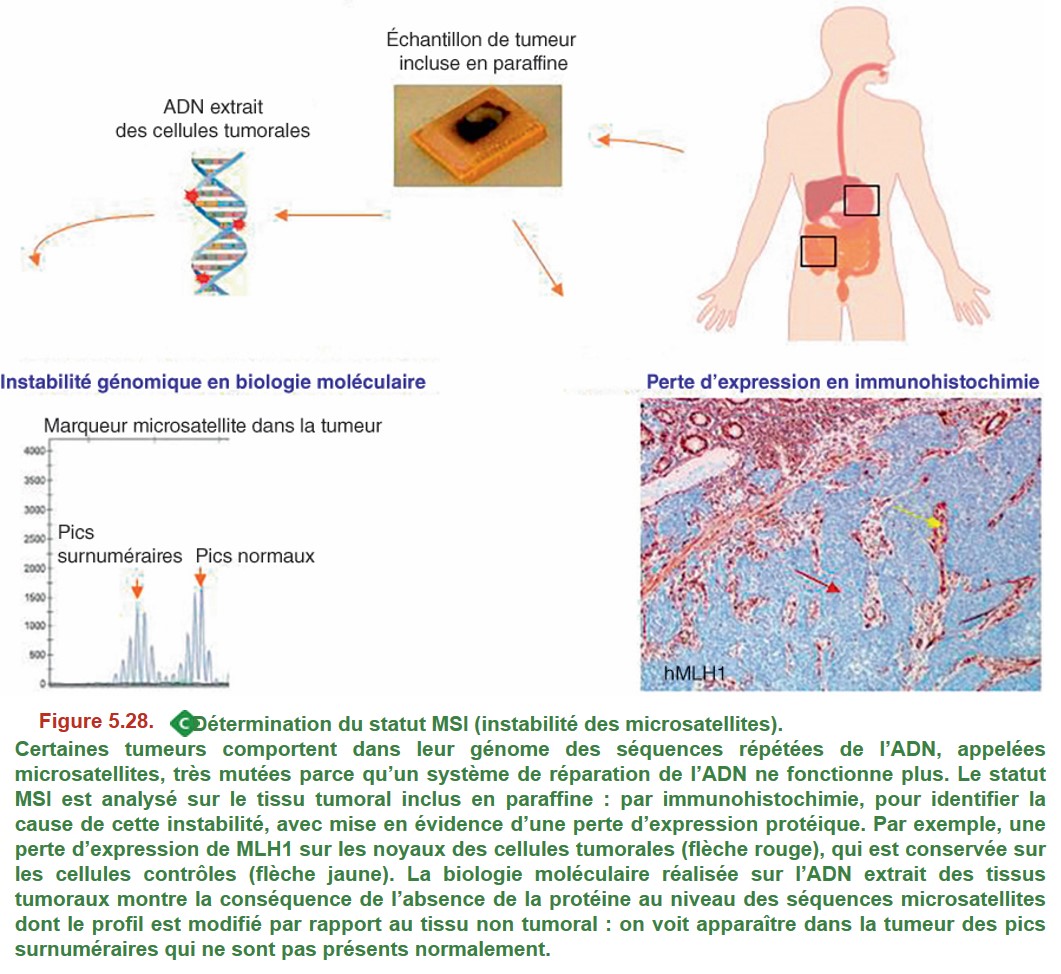
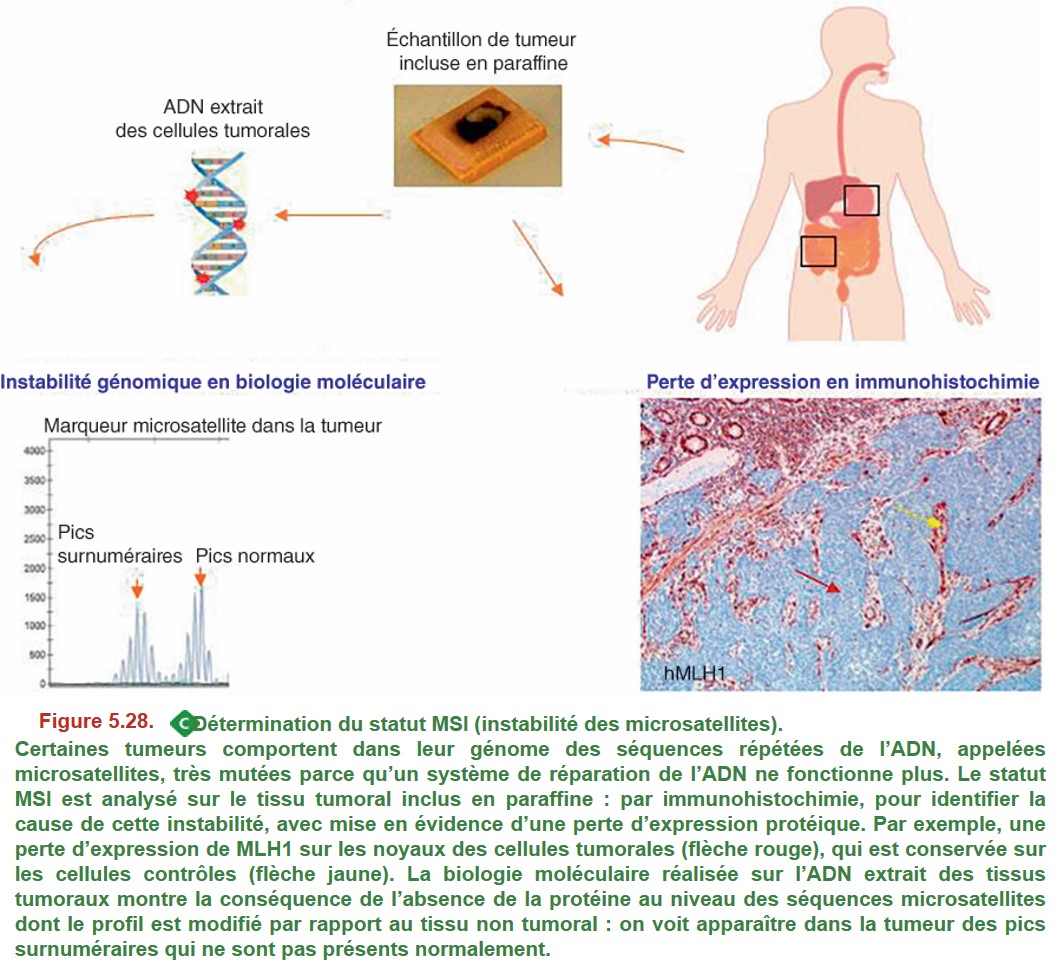
La détermination du statut MSI est recommandée de façon systématique (recommandations INCa 2021) pour son impact pronostique et théranostique (pronostic globalement meilleur des tumeurs MSI, réponse aux immunothérapies, sensibilité particulière aux chimiothérapies), et a également pour objectif d’orienter éventuellement le patient vers une consultation d’oncogénétique, à la recherche d’un syndrome de Lynch. Cette évaluation est faite par immunohistochimie et biologie moléculaire (Figure 5.28).
{kind=link}
Certaines tumeurs comportent dans leur génome des séquences répétées de l’ADN, appelées microsatellites, très mutées parce qu’un système de réparation de l’ADN ne fonctionne plus. Le statut MSI est analysé sur le tissu tumoral inclus en paraffine : par immunohistochimie, pour identifier la cause de cette instabilité, avec mise en évidence d’une perte d’expression protéique. Par exemple, une perte d’expression de MLH1 sur les noyaux des cellules tumorales (flèche rouge), qui est conservée sur les cellules contrôles (flèche jaune). La biologie moléculaire réalisée sur l’ADN extrait des tissus tumoraux montre la conséquence de l’absence de la protéine au niveau des séquences microsatellites dont le profil est modifié par rapport au tissu non tumoral : on voit apparaître dans la tumeur des pics surnuméraires qui ne sont pas présents normalement.
La recherche de phénotype MSI d’un cancer colorectal peut se faire par :
• immunohistochimie : par la mise en évidence d’une perte d’expression d’une ou plusieurs protéines MMR (principalement MLH1 ou MSH2) dans les cellules tumorales ;
• PCR à partir de l’ADN tumoral extrait du tissu fixé et inclus en paraffine restant dans les blocs tissulaires faits lors de l’exérèse de la tumeur et archivés au laboratoire d’anatomie patholo-gique.
4. Biologie moléculaire
En dehors de la recherche d’une instabilité des microsatellites qui peut être faite par biologie moléculaire, une recherche de mutations de KRAS et NRAS sur l’ADN tumoral extrait à partir des prélèvements anatomopathologiques est nécessaire en cas de maladie métastatique : la prescription d’anticorps monoclonaux anti-EGFR n’est possible qu’en l’absence de mutation activatrice de RAS.
Points clés
• Les cancers colorectaux sont le plus souvent des adénocarcinomes.
• L’adénocarcinome se développe en général à partir d’un adénome (tumeur épithéliale glandulaire bénigne) défini par son architecture (tubuleux, villeux ou tubulovilleux) et son degré de dysplasie (bas ou haut grade).
• Le risque de transformation carcinomateuse est majoré en cas d’adénome en dysplasie de haut grade, d’adénome > 1 cm, d’adénome d’architecture villeuse.
• Les adénomes se présentent sous la forme de polypes qui peuvent être réséqués par endoscopie dans la majorité des cas.
• Polype n’est pas synonyme d’adénome. Les autres polypes colorectaux très fréquents sont les polypes hyperplasiques qui ne dégénèrent pas.
• Le diagnostic d’adénocarcinome se fait par la réalisation d’une coloscopie totale avec biopsies multiples de la tumeur, et examen anatomopathologique.
• Lorsque le cancer est résécable, le traitement repose sur l’exérèse chirurgicale carcinologique de la tumeur et envoi de la pièce en anatomopathologie.
• L’examen anatomopathologique permet d’évaluer le pronostic et guide le traitement postopératoire.
Item 303 – Tumeurs de l’estomac
Auteurs : Florence Renaud, Dominique Wendum
I. Prérequis : histologie de la paroi gastrique
II. Les principales tumeurs gastriques
III. Adénocarcinome gastrique
IV. Autres tumeurs de l’estomac
Hiérarchisation des connaissances : Tableau 8
{kind=link}
I. Prérequis : histologie de la paroi gastrique
Cf. supra item 272 – Ulcère gastrique et duodénal. Gastrite.
II. Les principales tumeurs gastriques
III. Adénocarcinome gastrique
L’incidence de l’adénocarcinome de l’estomac est en diminution régulière en France. L’âge moyen de survenue est de 70 ans, avec une prédominance masculine, mais il existe des formes du sujet jeune, en particulier en cas de prédisposition génétique (cf. infra « Facteurs de risque et lésions tissulaires précancéreuses »). L’adénocarcinome est tumeur épithéliale glandulaire (adéno-) maligne (carcinome). Il peut être localisé dans la partie proximale de l’estomac (jonction œsogastrique, cardia et corps de l’estomac), ou dans la partie distale (antre, pylore).
A. Facteurs de risque et lésions tissulaires précancéreuses
Helicobacter pylori est le principal facteur de risque des cancers sporadiques. Il est à l’origine d’une infection chronique de la muqueuse gastrique associée à une réaction inflammatoire qui induit une gastrite chronique atrophique, état précancéreux. Les étapes suivantes de la carcinogenèse sont la métaplasie intestinale puis la dysplasie. HP serait responsable de 65 à 80 % des adénocarcinomes de l’estomac. Il est moins impliqué dans les localisations proximales (cardia). Les autres lésions tissulaires précancéreuses sont la gastrite atrophique auto-immune (ou maladie de Biermer), suivie de la même séquence métaplasie intestinale puis dysplasie, l’ulcère gastrique chronique, le polype gastrique adénomateux et la maladie de Ménétrier (gastropathie hypertro-phique). Il existe également des syndromes de prédisposition génétique, qui sont à l’origine de cancers habituellement chez le sujet jeune (< 40 ans) :
• syndrome de Lynch (gènes du système MMR) ;
• polypose adénomateuse familiale (gène APC) ;
• cancer gastrique diffus héréditaire (gène CDH1 en particulier).
B. Diagnostic des tumeurs de l’estomac
{kind=link}
C. Sous-types histologiques d’adénocarcinome
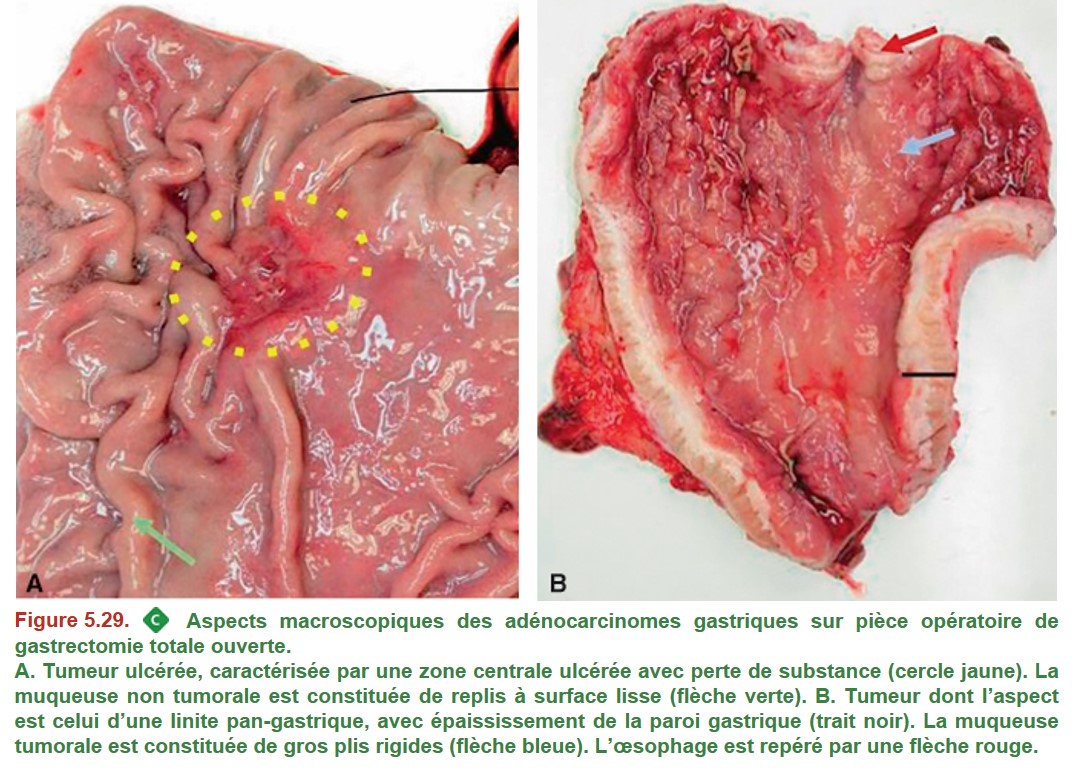
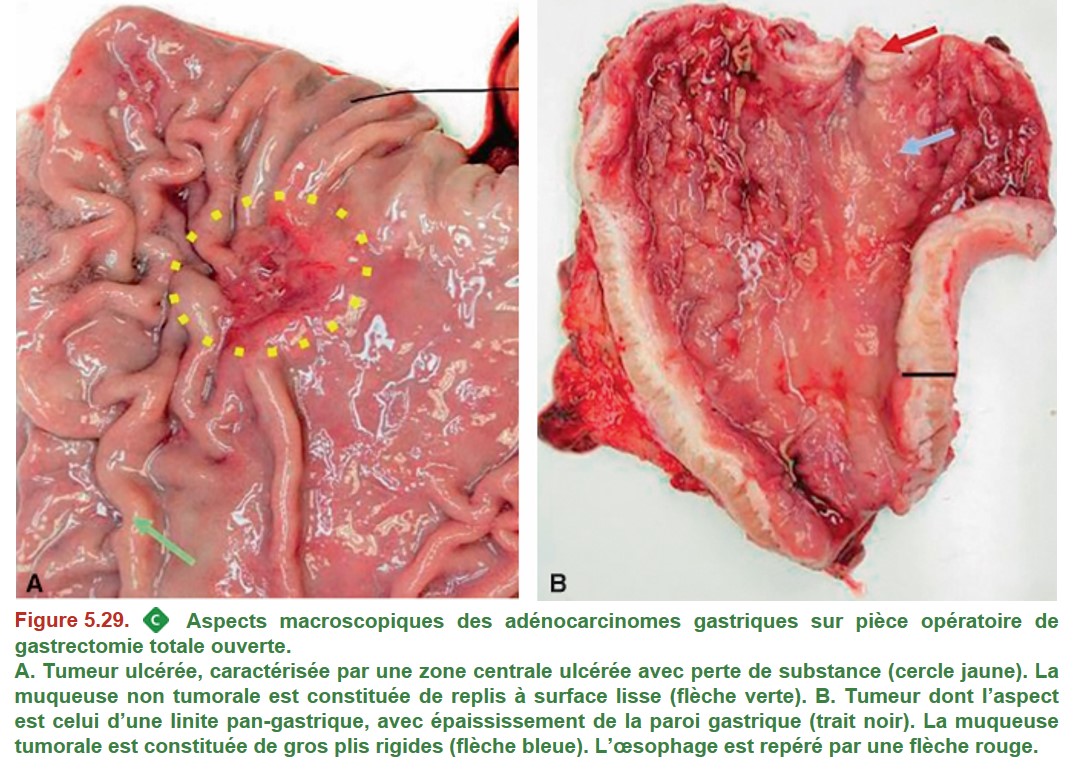
L’aspect macroscopique des adénocarcinomes est varié. Il s’agit le plus souvent d’une masse bourgeonnante ou d’un ulcère (Figure 5.29A). Rarement, leur aspect est celui d’une linite, caractérisé par un estomac à paroi épaissie et rigide (scléreuse), avec très peu d’anomalies visibles au ni-veau de la muqueuse (Figure 5.29B). Elle peut être difficile à diagnostiquer car les biopsies superficielles peuvent être négatives.
{kind=link}
{kind=link}
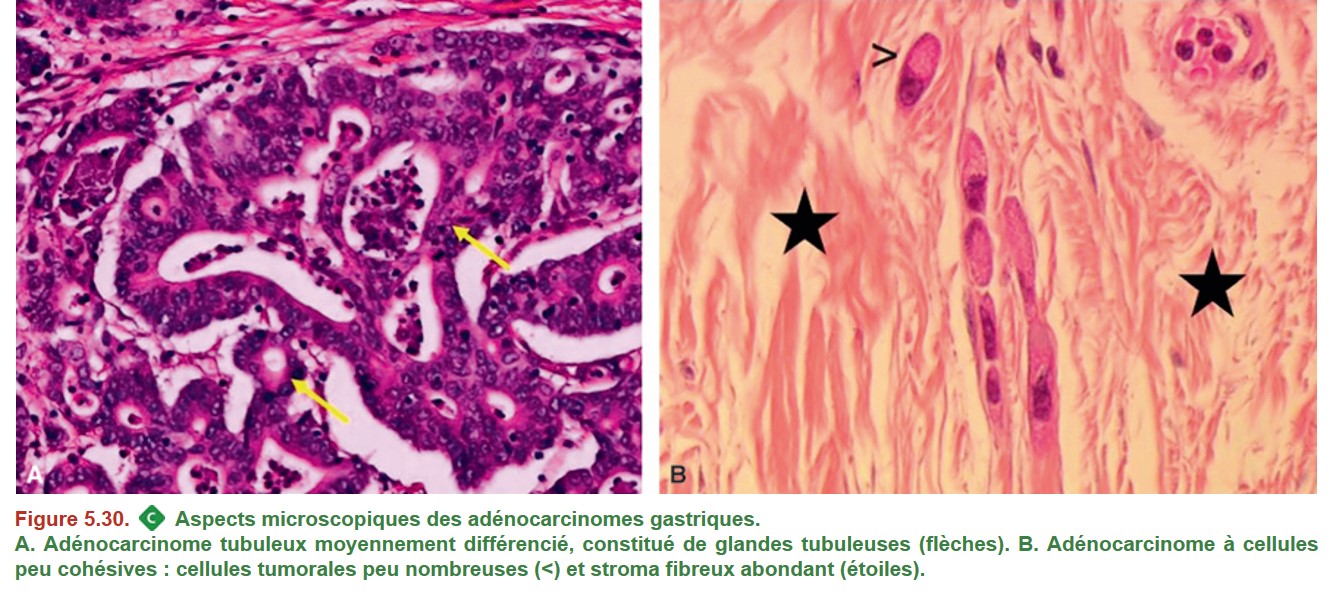
Il existe plusieurs classifications histologiques pour les adénocarcinomes : celle de l’OMS (adéno-carcinomes tubuleux/papillaires/mucineux/à cellules peu cohésives/autres) et la classification de Laurèn (adénocarcinomes de type intestinal/diffus/mixtes) (Figure 5.30). L’adénocarcinome de l’estomac est une tumeur très hétérogène et plusieurs sous-types au sein d’une même tumeur sont fréquemment associés. On précise le degré de différenciation des adénocarcinomes (bien, moyennement, peu).
{kind=link}
D. Carcinogenèse gastrique
Il s’agit d’un processus multifactoriel et multi-étapes. La gastrite chronique atrophique est le plus souvent associée à l’infection par HP, mais elle peut être d’origine auto-immune. Elle joue un rôle important dans la pathogénie de la majorité des adénocarcinomes de l’estomac de type intestinal. Elle constitue une condition favorisant la survenue du cancer gastrique et HP n’est qu’un des facteurs de risque impliqués. Les processus de carcinogenèse liés à l’adénocarcinome de type diffus à cellules peu cohésives sont moins bien connus hormis les rares formes génétiques familiales de cancer gastrique liées au gène CDH1.
E. Évolution naturelle
La survie à 5 ans chez le patient opéré (maladie localisée) est de l’ordre de 40 %. En cas de métastase, la durée de vie médiane est de 12 à 16 mois (grandes variations individuelles).
IV. Autres tumeurs de l’estomac
A. Tumeurs stromales gastro-intestinales (GIST)
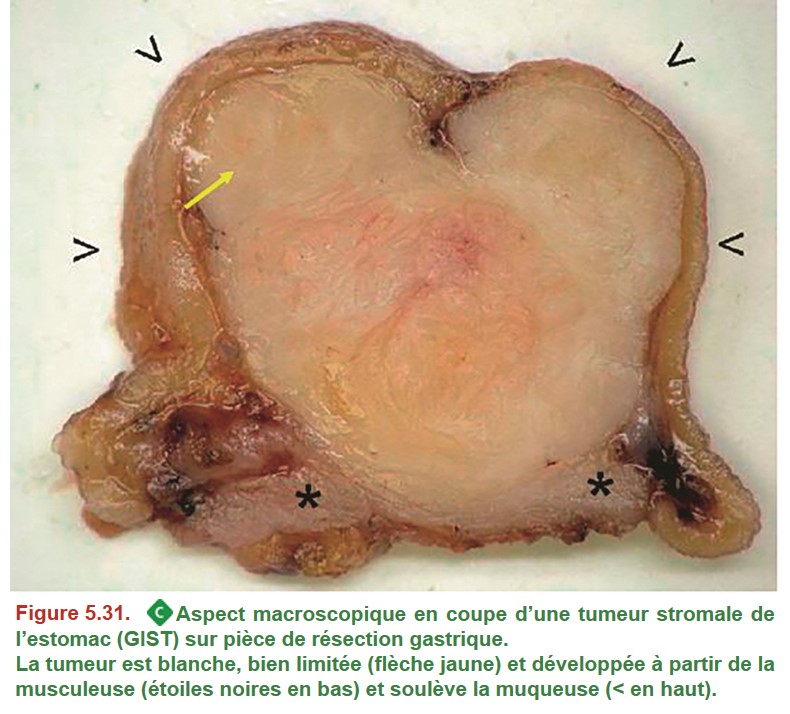
Les GIST sont des tumeurs mésenchymateuses (famille des sarcomes), qui se développent fréquemment dans l’estomac ou l’intestin grêle, au niveau de la musculeuse.
{kind=link}
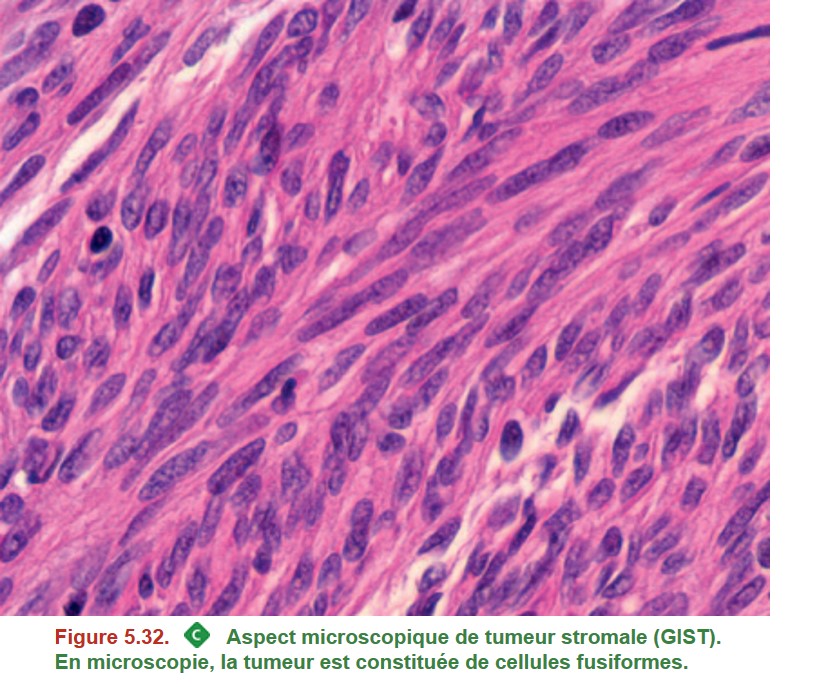
La tumeur est blanche, bien limitée (flèche jaune) et développée à partir de la musculeuse (étoiles noires en bas) et soulève la muqueuse (< en haut). Les biopsies endoscopiques sont souvent négatives car la tumeur est profonde. Le diagnostic anatomopathologique s’appuie sur la morphologie qui met en évidence des cellules allongées (= fusiformes) (Figure 5.32) et l’étude immunohistochimique (obligatoire pour le diagnostic) montrant une expression de la protéine KIT (CD117) et/ou DOG-1 par les cellules tumorales. Il existe souvent une mutation du gène KIT dans la tumeur.
{kind=link}
B. Lymphomes primitifs gastriques
Ils représentent environ 5 % des tumeurs de l’estomac. Le lymphome primitif de l’estomac le plus fréquent est le lymphome B à petites cellules du MALT. Ces lymphomes extraganglionnaires naissent de la zone marginale du MALT, en lien avec l’infection chronique à HP dans plus de 90 % des cas. Ils sont en majorité associés à un faible degré de malignité.
Le diagnostic se fait par endoscopie œsogastroduodénale avec biopsies multiples de la lésion gastrique et examen anatomopathologique des biopsies.
L’examen anatomopathologique :
• fait le diagnostic de lymphome ;
• précise le type de lymphome suivant la classification de l’OMS en vigueur ;
• précise la présence ou non d’HP.
Le diagnostic de lymphome nécessite de l’immunohistochimie et assez souvent de la biologie mo-léculaire.
C. Tumeurs neuroendocrines gastriques
Les cellules neuroendocrines du tube digestif font partie du système endocrine diffus. Ce sont des cellules épithéliales dispersées au sein des épithéliums de revêtement. Elles sont dites neuroendocrines car elles sécrètent des amines ou peptides hormonaux (endocrines) et expriment également des marqueurs nerveux (CD-56 ou N-CAM, synaptophysine par exemple). Dans l’estomac, les cellules neuroendocrines sécrètent principalement de l’histamine, de la gastrine ou de la sérotonine.
Dans l’estomac, les tumeurs neuroendocrines peuvent être :
• associées à une gastrite chronique atrophique fundique auto-immune (physiopathologie : la destruction des cellules pariétales entraîne une achlorhydrie puis une hypergastrinémie réactionnelle qui stimule la prolifération des cellules ECL [cellules endocrines situées dans le fundus]) ;
• associées à une néoplasie endocrinienne multiple de type 1 – syndrome de Zollinger-Ellison (gastrinome dans le cadre de la NEM-1 stimulant aussi la prolifération des cellules ECL de l’estomac).
Ces tumeurs sont de bon pronostic ;
• sporadiques, de moins bon pronostic.
Il existe un système de grade et un système de stadification spécifiques pour les tumeurs neuroendocrines, différents de ceux des autres carcinomes.
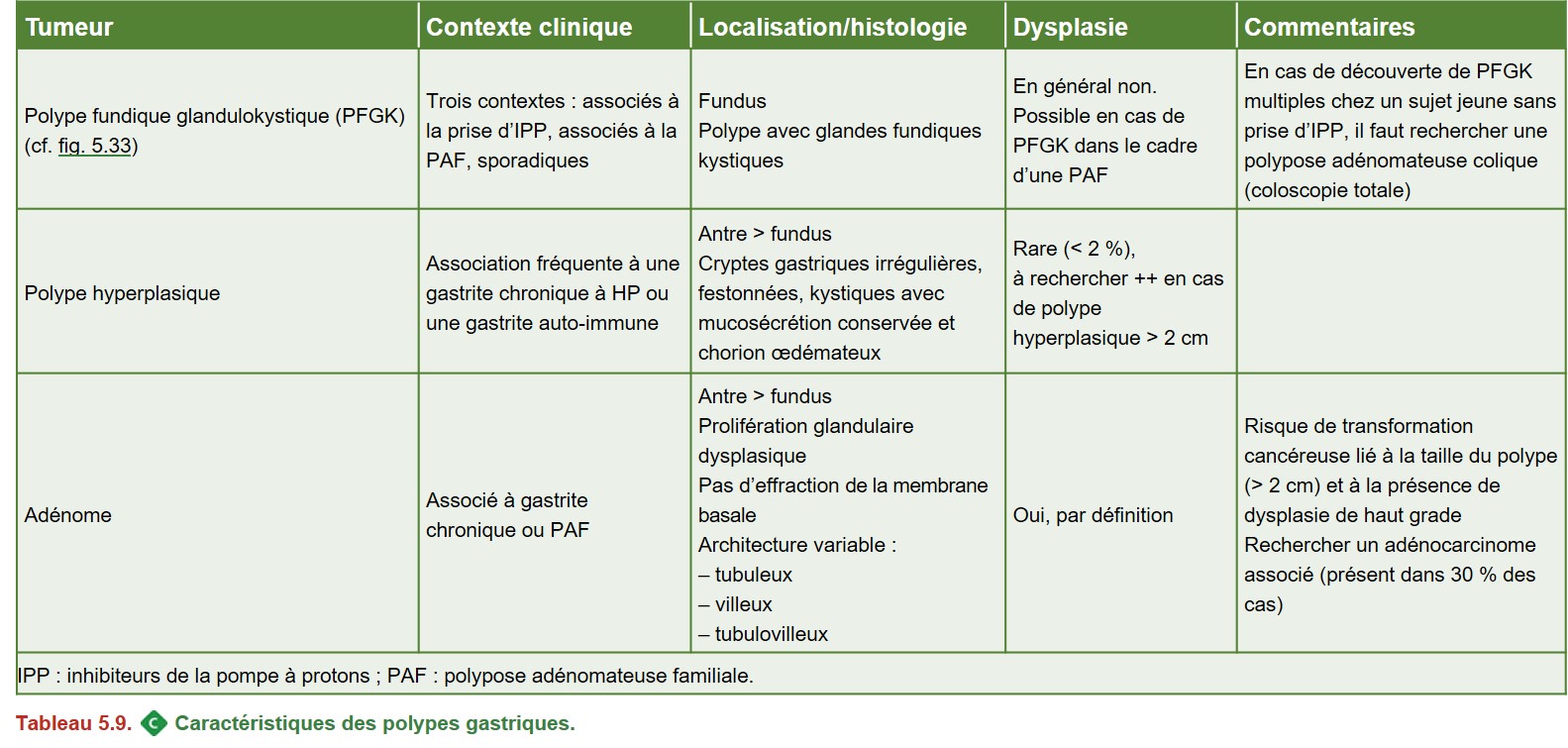
D. Polypes gastriques
Les plus fréquents au niveau de l’estomac sont :
• les polypes glandulokystiques : de loin les plus fréquents, situés dans le fundus ;
• les polypes hyperplasiques : deuxièmes en fréquence, le plus souvent dans l’antre ;
• les adénomes.
{kind=link}
Les polypes gastriques sont biopsiés pour en préciser la nature.
Un geste de résection (polypectomie ou mucosectomie) est recommandé en cas :
• de polype hyperplasique > 5 mm ;
• d’adénome.
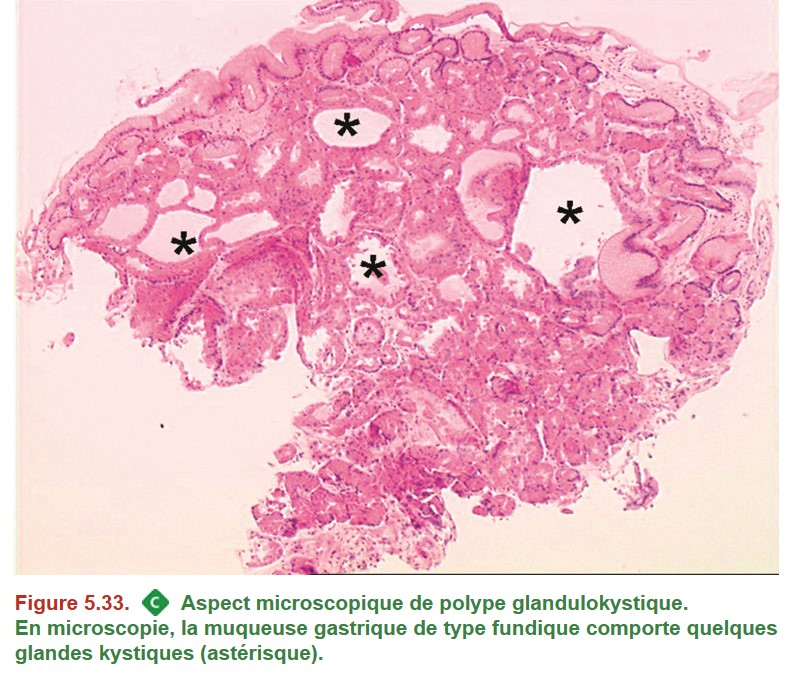
Les polypes fundiques glandulokystiques (Figure 5.33) peuvent être laissés en place, sauf en cas de contexte de PAF, auquel cas ils doivent être retirés s’ils mesurent plus de 1 cm.
{kind=link}
Il existe d’autres polypes gastriques, plus rares : polypes hamartomateux, polypes fibro-inflammatoires, xanthome, hétérotopie pancréatique, etc.
Points clés
• L’adénocarcinome est la tumeur maligne gastrique la plus fréquente.
• Les lésions tissulaires précancéreuses sont la gastrite chronique atrophique, l’ulcère gastrique chronique, le polype gastrique adénomateux et la maladie de Ménétrier (gastropathie hypertrophique).
• Le diagnostic se fait par endoscopie avec biopsies multiples de la lésion (8 à 10), et examen anatomo-pathologique.
• La linite gastrique est une forme particulière d’adénocarcinome : la paroi est épaissie et rigide avec peu d’anomalies au niveau de la muqueuse ; la prolifération adénocarcinomateuse est souvent à cellules peu cohésives. Elle peut être difficile à diagnostiquer car les biopsies superficielles peuvent être négatives.
• Le cancer superficiel de l’estomac est un cancer T1 (invasion de la muqueuse ou de la sous-muqueuse).