
CHAPITRE IX - Hématologie
Auteur : Pierre Reimbold
Plan :
• Item 318 – Leucémies lymphoïdes chroniques
• Item 319 – Lymphomes malins
• Item 320 – Myélome multiple des os
• Item 220 – Adénopathie superficielle de l’adulte et de l’enfant
Item 318 – Leucémies lymphoïdes chroniques
I. Définition
II. Épidémiologie
III. Présentations et manifestations cliniques
IV. Diagnostic
V. Pronostic
VI. Syndrome de Richter
VII. Diagnostics différentiels
Hiérarchisation des connaissances : tableau 1
{kind=link}
I. Définition
II. Épidémiologie
III. Présentations et manifestations cliniques
Cette prolifération tumorale de petits lymphocytes B se manifeste par :
• une hyperlymphocytose dans le sang ;
• parfois associée à des adénopathies et une splénomégalie (inconstantes) ;
• des cytopénies liées à l’infiltration de la moelle hématopoïétique ou à des complications auto-immunes ;
• une immunodépression à l’origine d’infections à répétition (hypogammaglobulinémie) ou d’une augmentation de l’incidence des cancers solides (cutanés en particulier). Néanmoins, le plus souvent, la découverte est fortuite avec un hémogramme retrouvant une hyperlymphocytose supérieure à 5 G/L chez un patient asymptomatique (d’après la classification OMS 2022).
IV. Diagnostic
A. Formes leucémiques
Le diagnostic est suspecté sur l’hémogramme retrouvant :
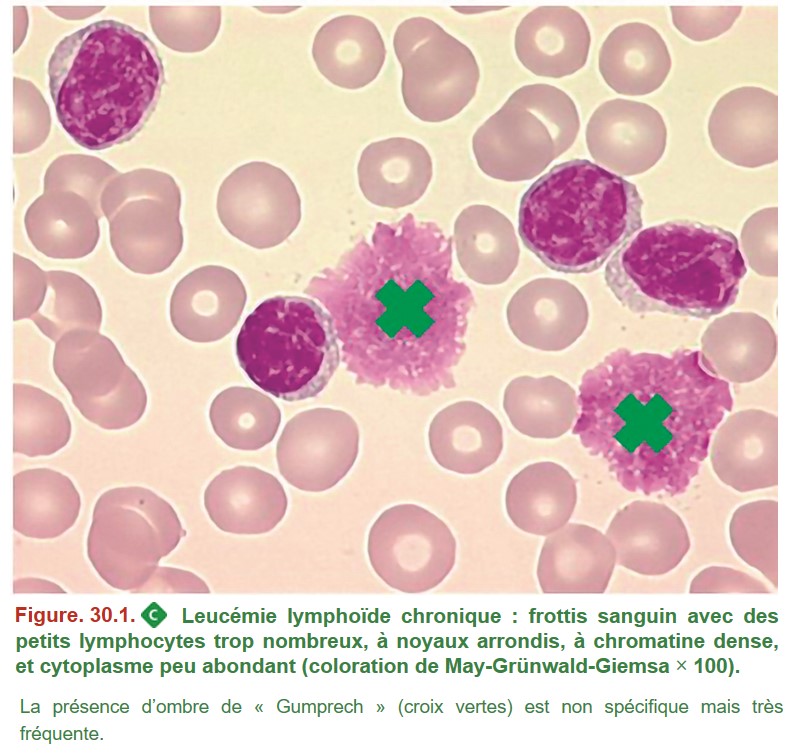
• une hyperlymphocytose durable > 5 G/L ;
• avec un frottis sanguin montrant des petits lymphocytes d’aspect mature (figure. 30.1), trop nombreux, à la chromatine dense et souvent associés à des « ombres de Gumprecht », qui correspondent à des lymphocytes rompus lors de l’étalement du frottis sanguin.
{kind=link}
B. Formes non leucémiques
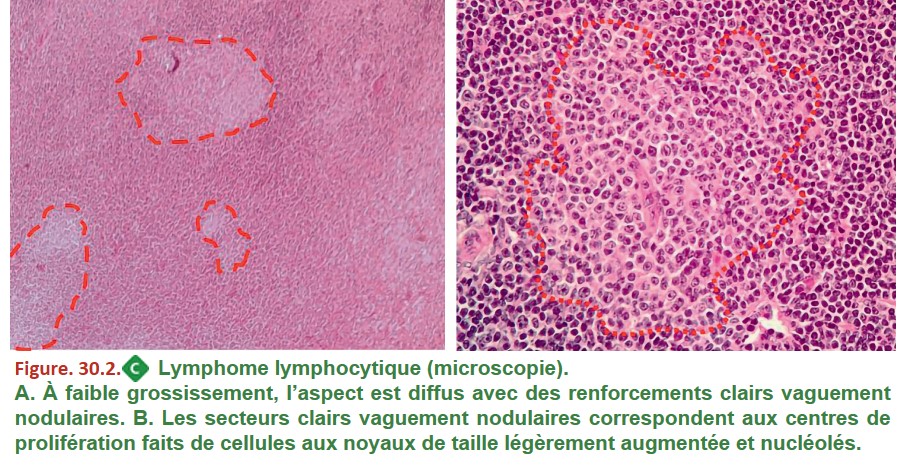
Ces formes (lymphomes lymphocytiques) ne s’accompagnant pas ou peu de lymphocytes circulants ; le diagnostic ne peut pas se faire sur une analyse du sang circulant. Il repose sur l’analyse anatomopathologique d’une biopsie ostéomédullaire ou d’une biopsie de ganglion. Dans les deux cas, le tissu est le siège d’une infiltration tumorale d’architecture diffuse, constituée de petits lymphocytes B d’aspect mature avec des zones claires appelées centres de prolifération contenant des cellules de plus grande taille (figure. 30.2).
{kind=link}
Remarque :
V. Pronostic
VI. Syndrome de Richter
Il doit être suspecté en cas :
• de sueurs nocturnes ;
• de fièvre au long cours ;
• d’amaigrissement ;
• d’augmentation des LDH ;
• d’apparition d’une adénopathie volumineuse, asymétrique, douloureuse ;
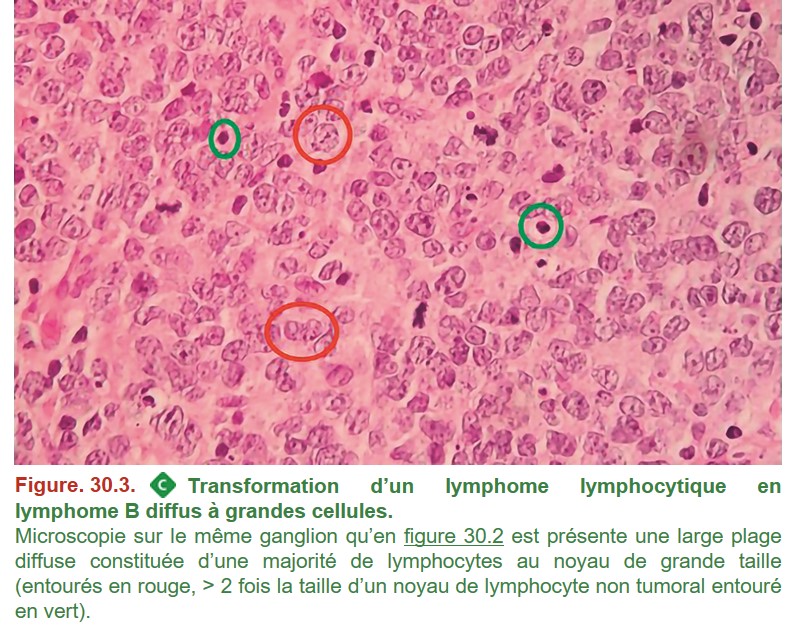
• d’apparition, sur un TEP-scanner (tomographie par émission de positons), d’un territoire ganglionnaire présentant des SUVmax (standardized uptake values) plus élevées que les autres territoires ganglionnaires.
La confirmation diagnostique nécessite une biopsie ganglionnaire qui retrouve une prolifération diffuse de lymphocytes B de grande taille (figure. 30.3).
{kind=link}
VII. Diagnostics différentiels
• les formes leucémiques de certains lymphomes :
– lymphome à cellules du manteau,
– lymphome de la zone marginale,
– lymphome lymphoplasmocytaire ;
• la leucémie à tricholeucocyte ;
• certaines hémopathies lymphoïdes T (leucémie prolymphocytaire T notamment) ;
• pour les lymphomes lymphocytiques : tous les autres lymphomes B à petites cellules.
Points clés
• La LLC correspond à une prolifération tumorale de petits lymphocytes B matures avec une lymphocytose supérieure à 5 G/L.
• Le diagnostic de LLC repose sur l’immunophénotypage des lymphocytes circulants en laboratoire d’hématologie par cytométrie en flux.
• Le diagnostic est anatomopathologique en cas d’absence de passage sanguin des cellules tumorales (lymphome lymphocytique). Il se fait alors sur une biopsie médullaire ou une biopsie de ganglion.
• Le diagnostic de transformation d’une LLC en lymphome à grandes cellules (syndrome de Richter) est anatomopathologique et nécessite une biopsie ganglionnaire.
Item 319 – Lymphomes malins
I. Définition d’un lymphome malin
II. Principes de la classification des lymphomes selon l’OMS 2017
III. Épidémiologie des lymphomes non hodgkiniens
IV. Diagnostic d’un lymphome
V. Principaux lymphomes non hodgkiniens B
VI. Lymphomes hodgkiniens
Hiérarchisation des connaissances : tableau 2
{kind=link}
I. Définition d’un lymphome malin
II. Principes de la classification des lymphomes
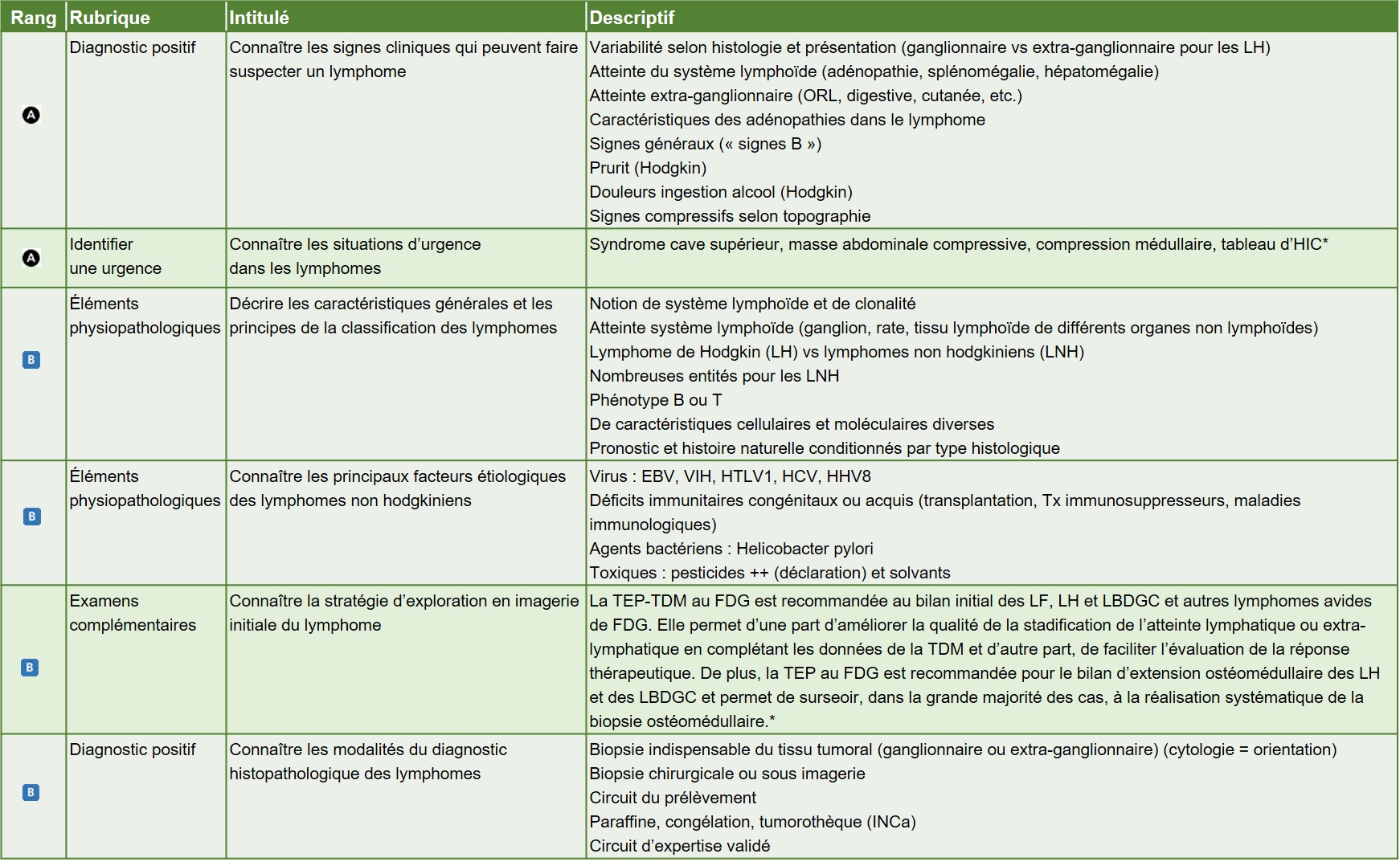
La classification des lymphomes distingue les lymphomes hodgkiniens, qui sont des lymphomes B ayant une présentation clinique et microscopique particulière, des autres lymphomes alors dits lymphomes non hodgkiniens. Cette classification repose sur la confrontation d’un ensemble de données permettant de rapprocher la cellule lymphomateuse d’une contrepartie normale dans la voie de différenciation du lymphocyte B ou T (figure. 31.1).
{kind=link}
Elle permet ainsi d’identifier des lymphomes d’évolution et de pronostic différents avec :
• des lymphomes dits « agressifs » (ex : lymphome B diffus à grandes cellules, lymphome de Burkitt), d’évolution rapide nécessitant un traitement par chimiothérapies intensives ;
• des lymphomes dits « indolents » (ex : lymphome lymphocytique, lymphome folliculaire), d’évolution plus lente, qui ne sont pas éradicables à l’heure actuelle, et dont l’histoire clinique alterne entre des phases de traitement et des phases de rémission.
La lymphe est acheminée au ganglion lymphatique par les vaisseaux lymphatiques afférents. La lymphe contient les cellules présentatrices d’antigènes qui rejoignent les follicules lymphoïdes situés dans le cortex. Elles participent ainsi au développement de l’immunité adaptative au sein des centres germinatifs. Les lymphocytes B naïfs du manteau migrent dans le centre germinatif afin d’être sélectionnés pour donner lieu à des lymphocytes B mémoires et des plasmocytes qui quittent le centre germinatif en passant par la zone marginale. La plupart des lymphomes B peuvent être rattachés à une cellule d’origine supposée correspondre à un stade de maturation du lymphocyte B, en fonction des différents marqueurs de différenciation exprimés par la cellule tumorale.
III. Épidémiologie des lymphomes non hodgkiniens
Plus rarement, ils peuvent être favorisés par :
• un agent infectieux (EBV, HTLV1 [human T lymphotropic virus 1], HHV8 [human herpes virus 8]) ;
• un contexte d’immunodéficience (ex : traitements immunosuppresseurs dans un contexte de greffe d’organe, VIH) ;
• une inflammation chronique, qu’elle soit infectieuse (ex : gastrite chronique à Helicobacter pylori) ou auto-immune (ex : syndrome de Gougerot-Sjögren) ;
• des facteurs environnementaux (ex : exposition professionnelle aux pesticides).
IV. Diagnostic d’un lymphome
Il est pluridisciplinaire et repose sur la confrontation des données :
• cliniques (présentation, localisation, âge, terrain) ;
• anatomopathologiques :
– morphologiques (architecture de la prolifération tumorale et aspect cytologique),
– immunohistochimiques (caractérisation des protéines exprimées par les cellules tumorales = marqueurs de différenciation) ;
• cytogénétiques (ex : translocations chromosomiques) et moléculaires (mutations récurrentes de certains gènes).
Ainsi, le diagnostic de lymphome est le plus souvent porté par une biopsie à l’aiguille ou par une biopsie chirurgicale d’un ganglion ou d’une localisation extra-ganglionnaire du lymphome.
Le prélèvement doit être :
• de taille suffisante (privilégier une exérèse ganglionnaire à une biopsie à l’aiguille) ;
• acheminé à l’état frais au laboratoire d’anatomie et cytologie pathologiques.
Ce prélèvement frais (sans fixateur) fait obligatoirement l’objet :
• d’une congélation ;
• d’une fixation en formol pour examen morphologique et étude immunohistochimique ;
•
En fonction du matériel disponible et des diagnostics suspectés, le prélèvement frais peut aussi faire l’objet d’une mise en suspension cellulaire pour immunophénotypage en cytométrie de flux, et/ou d’une mise en culture pour étude en cytogénétique conventionnelle : caryotype et étude en FISH sur chromosomes métaphasiques.
• l’analyse morphologique ;
• l’analyse immunohistochimique (étude de l’expression des marqueurs de différenciation par les cellules tumorales à l’aide d’anticorps spécifiques) ;
• et parfois l’hybridation in situ (recherche de génome viral comme l’EBV par exemple) et/ou analyse en FISH pour recherche d’anomalies cytogénétiques sur coupes de tissu fixées.
Le fragment congelé sert éventuellement pour :
• une recherche de clonalité (recherche d’un réarrangement clonal des gènes IGH ou du TCR) ;
• la recherche d’anomalies moléculaires par techniques de biologie moléculaire (NGS par exemple).
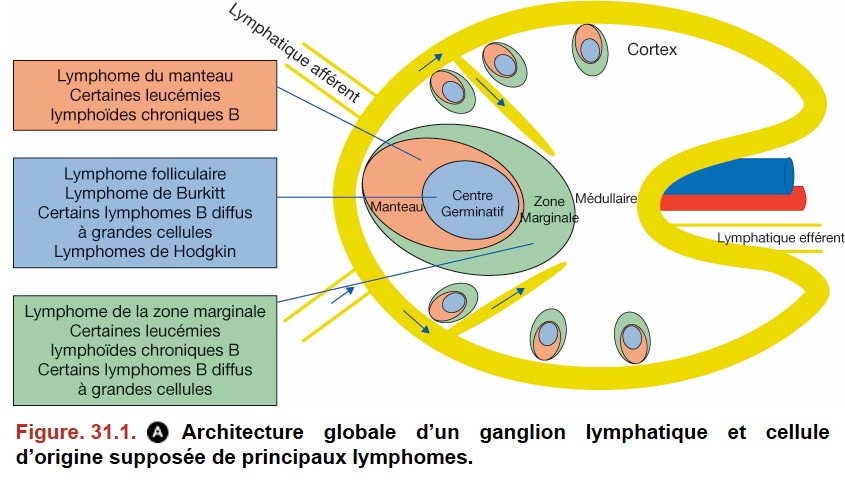
V. Principaux lymphomes non hodgkiniens B
{kind=link}
{kind=link}
{kind=link}
Les principaux lymphomes B à petites cellules sont :
• le lymphome folliculaire ;
• le lymphome lymphocytique/leucémie lymphoïde chronique ;
• le lymphome de la zone marginale ;
• le lymphome du manteau ;
• le lymphome lymphoplasmocytaire
Hormis le lymphome du manteau, les lymphomes à petites cellules sont indolents et leur évolution alterne entre des périodes de traitement et des périodes de rémission. Les principaux lymphomes non hodgkiniens sont présentés dans la figure 31.3.
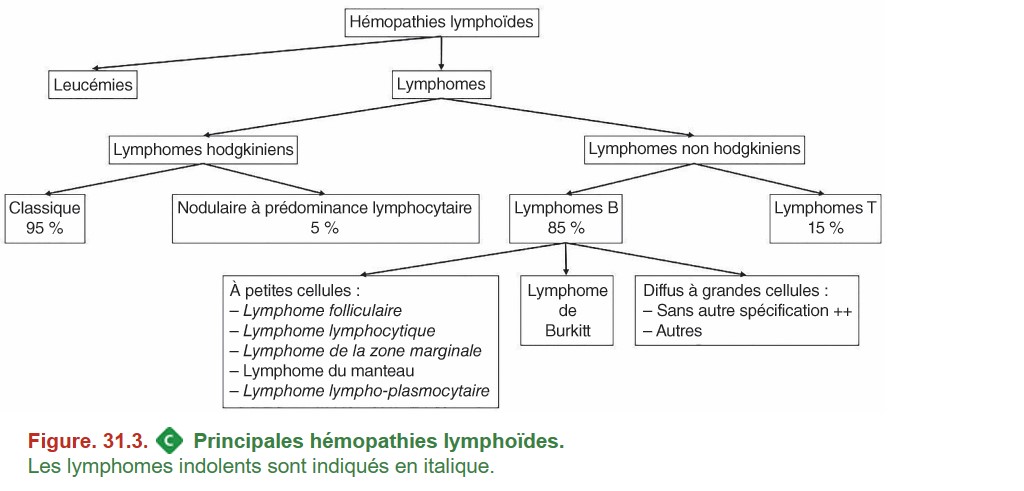{kind=link}
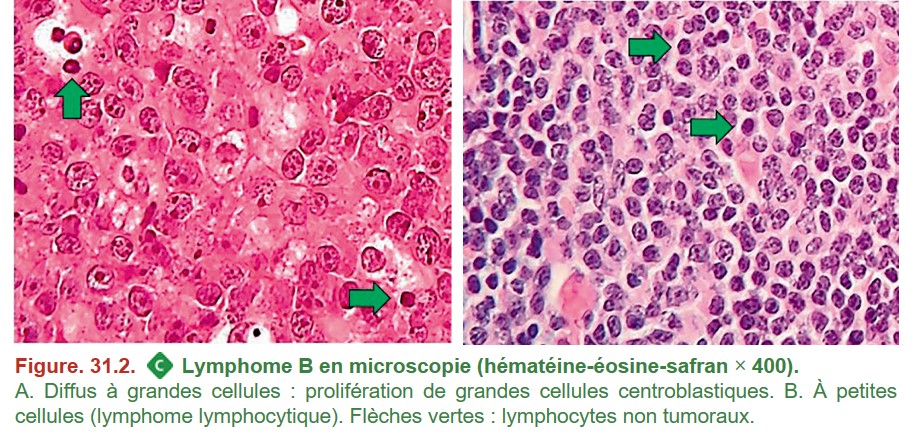
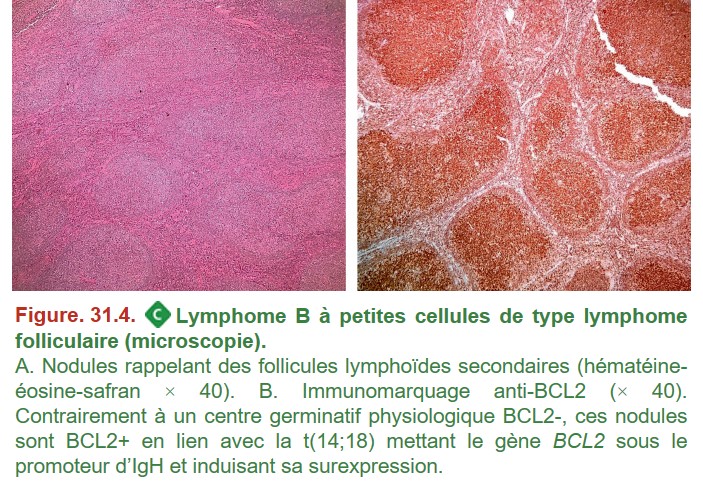
L’architecture de la prolifération lymphomateuse ainsi que l’aspect des cellules permettent de s’orienter vers une ou plusieurs hypothèses diagnostiques. Afin d’étayer le diagnostic, des techniques complémentaires sont réalisées pour rapprocher la cellule tumorale d’une contrepartie normale dans la voie de différenciation lymphocytaire (immunohistochimie) et d’identifier des anomalies moléculaires associées à un lymphome (FISH, cytogénétique, biologie moléculaire) (figure. 31.4).
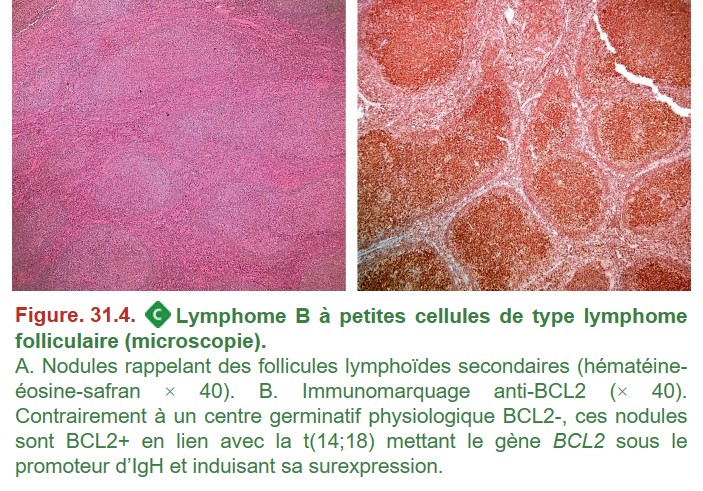{kind=link}
VI. Lymphomes hodgkiniens
Pour les lymphomes de Hodgkin, la classification de l’OMS 2022 distingue deux entités clinicopathologiques :
• le lymphome de Hodgkin classique (95 % des cas) avec quatre variants histologiques (scléronodulaire, à cellularité mixte, riche en lymphocytes, avec déplétion lymphocytaire) et dont la forme scléronodulaire est la plus fréquente (70 % des cas) ;
• le lymphome de Hodgkin nodulaire à prédominance lymphocytaire, encore appelé paragranulome de Poppema-Lennert (5 % des cas), pouvant évoluer vers un lymphome B diffus à grandes cellules.
Le diagnostic histopathologique repose sur la reconnaissance (figure. 31.5) :
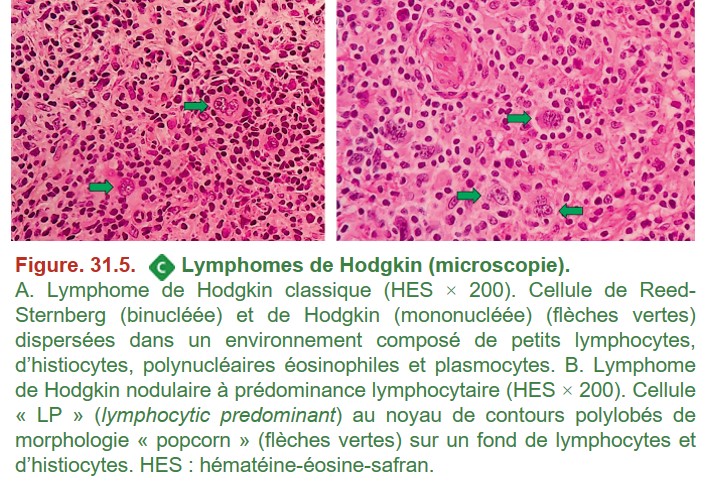{kind=link}
• de cellules de Hodgkin et de Reed-Sternberg pour le lymphome de Hodgkin classique ;
• ou des cellules de type LP (lymphocytic predominant cells appelées parfois « cellules pop-corn ») pour le lymphome de Hodgkin nodulaire à prédominance lymphocytaire ;
• le tout dans un environnement tumoral approprié.
Remarque :
Points clés
• Les lymphomes malins sont un groupe de maladies résultant de la prolifération maligne de cellules lymphoïdes B, T ou NK à différents stades de différenciation.
• Le diagnostic de lymphome est pluridisciplinaire et repose sur la confrontation des données cliniques, biologiques, morphologiques, immunophénotypiques, moléculaires et cytogénétiques.
• Sur le plan anatomopathologique, leur diagnostic est parfois complexe, ce pour quoi il existe un réseau national de double lecture systématisée de chaque nouveau diagnostic de lymphome.
• Le prélèvement tissulaire pour diagnostic doit être :
– de taille suffisante (privilégier une exérèse ganglionnaire à une biopsie à l’aiguille) ;
– acheminé à l’état frais au laboratoire d’anatomie et cytologie pathologiques.
• Ce prélèvement frais (sans fixateur) fait obligatoirement l’objet d’une fixation par du formol tamponné et d’une congélation.
• On distingue les lymphomes de Hodgkin des lymphomes non hodgkiniens B ou T.
• Parmi les lymphomes non hodgkiniens, 85 % sont de phénotype B. Les plus fréquents sont le lymphome diffus à grandes cellules B (35 %) et le lymphome folliculaire (22 %).
• Pour les lymphomes hodgkiniens, le diagnostic histopathologique repose sur l’identification de cellules de Hodgkin/Reed-Sternberg pour le lymphome de Hodgkin classique et de cellules dites de « type LP » pour le lymphome de Hodgkin nodulaire à prédominance lymphocytaire (paragranulome de Poppema), au sein d’un environnement tumoral approprié.
Pour en savoir plus : Société française d’hématologie. Hématologie, 4e édition. Issy-Les-Moulineaux : Elsevier Masson ; 2021.
Item 320 – Myélome multiple des os
I. Définition
II. Présentations cliniques des néoplasmes plasmocytaires
III. Diagnostic
IV. Diagnostics différentiels
Hiérarchisation des connaissances : tableau 3
{kind=link}
I. Définition
II. Présentations cliniques des néoplasmes plasmocytaires
A. Plasmocytome solitaire
Le plasmocytome solitaire correspond à une prolifération plasmocytaire maligne localisée au niveau osseux ou extra-osseux (par exemple au niveau du tractus digestif) associée à la présence ou non d’une immunoglobuline monoclonale. Celui-ci peut induire des symptômes en rapport avec le syndrome de masse tumorale. Le myélogramme est normal, et le diagnostic de plasmocytome repose sur la ponction-biopsie de la lésion. Le diagnostic de plasmocytome solitaire ne peut quant à lui être retenu qu’après un bilan clinique, biologique et d’imagerie (IRM corps entier et/ou TEP-scanner) ne mettant pas en évidence d’autre lésion osseuse. Il existe une évolution possible vers un myélome estimée aux alentours de 50-70 % à 10 ans pour les plasmocytomes osseux et beaucoup plus rare (estimée < 10 %) pour les plasmocytomes extra-osseux. Ils sont le plus souvent traités par radiothérapie suivie d’une surveillance régulière clinicobiologique et radiologique.
B. Myélome multiple
La forme diffuse correspond au myélome multiple, dont le syndrome tumoral est principalement caractérisé par les atteintes osseuses. Il n’y a le plus souvent pas d’adénopathie périphérique ni d’hépatosplénomégalie, mais il peut exister un passage sanguin des plasmocytes tumoraux pouvant à l’extrême réaliser un tableau de leucémie à plasmocytes (plasmocytose > 2 G/L ou > 20 % des leucocytes circulants).
III. Diagnostic
A. Myélogramme
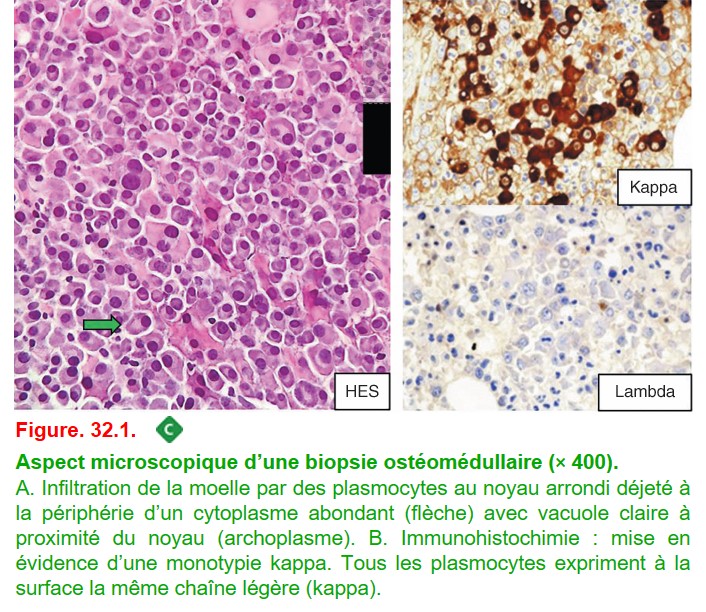
Le myélogramme par ponction sternale est un examen cytologique pris en charge par les laboratoires d’hématologie. L’aspiration de moelle hématopoïétique est étalée sur des lames qui sont colorées par le May-Grünewald-Giemsa (MGG) pour le diagnostic cytologique. Le diagnostic de myélome repose alors sur la mise en évidence de plasmocytes, représentant plus de 10 % de l’ensemble des éléments nucléés visualisés.
B. Biopsie ostéomédullaire ou biopsie osseuse radioguidée
{kind=link}
{kind=link}
IV. Diagnostics différentiels
La découverte d’une gammapathie monoclonale doit en premier lieu faire évoquer le diagnostic de myélome multiple et de ses précurseurs asymptomatiques : gammapathie monoclonale de signification indéterminée (MGUS) et myélome indolent. Cependant, d’autres entités peuvent s’accompagner d’une immunoglobuline monoclonale et constituent ainsi des diagnostics différentiels :
• la maladie de Waldenström (lymphome lymphoplasmocytaire) ;
• d’autres lymphomes B de bas grade : le plus fréquemment associé à une immunoglobuline monoclonale étant le lymphome de la zone marginale ;
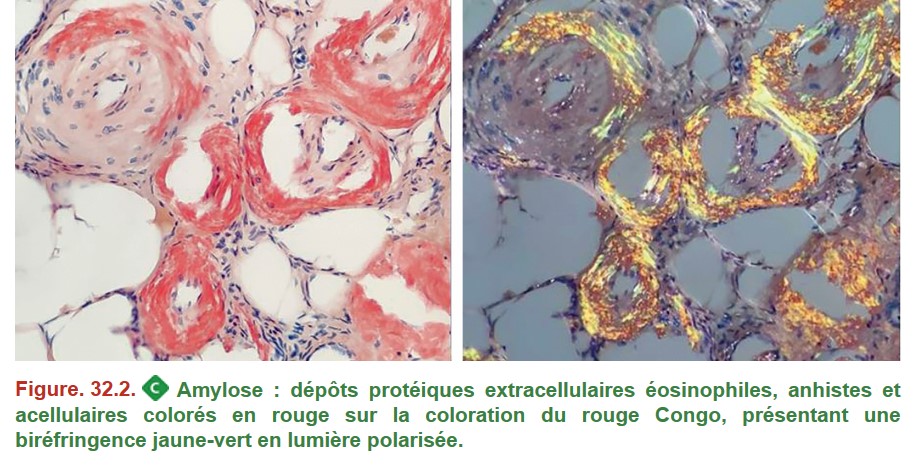
• l’amylose AL, correspondant à des dépôts amyloïdes d’immunoglobulines au sein de différents organes parfois par un clone plasmocytaire très minoritaire et non détectable.
Points clés
• Le myélome multiple est une hémopathie lymphoïde B mature du sujet âgé, caractérisée par une prolifération plasmocytaire maligne se développant majoritairement au sein de la moelle hématopoïétique.
• Le diagnostic de myélome repose le plus souvent sur le myélogramme avec présence de plasmocytes représentant plus de 10 % des éléments nucléés.
Item 220 – Adénopathie superficielle de l’adulte et de l’enfant
I. Prérequis
II. Démarche diagnostique
III. Intérêt et valeur des différents prélèvements ganglionnaires pour examen anatomopathologique
Hiérarchisation des connaissances : tableau 4
{kind=link}
I. Prérequis : Définition
Une adénopathie est un ganglion lymphatique présentant des caractéristiques cliniques et/ou radiologiques suspectes.
Le terme d’adénomégalie quant à lui ne préjuge pas du caractère pathologique ou non d’un ganglion de taille augmentée, supérieure à 1 cm.
II. Démarche diagnostique
• une réaction immunitaire normale (ganglion réactionnel secondaire à une infection dans le territoire de drainage par exemple) ;
• une infection systémique (mononucléose infectieuse, infection à VIH, etc.) ;
• une infection directe du ganglion (tuberculose ganglionnaire, maladie des griffes du chat, etc.) ;
• une pathologie inflammatoire dysimmunitaire (sarcoïdose, lupus, etc.) ;
• une localisation tumorale maligne :
– hémopathie lymphoïde (lymphome de Hodgkin, lymphome non hodgkinien),
– métastase ganglionnaire d’un autre type de cancer (carcinome, mélanome).
III. Intérêt et valeur des différents prélèvements ganglionnaires pour examen anatomopathologique
A. Cytoponction ganglionnaire
Elle est pratiquée à l’aide d’une aiguille fine permettant d’obtenir des cellules qui peuvent être étalées par frottis sur lames qui sont séchées à l’air avant d’être colorées. En plus de l’analyse cytologique mise sur lame, la cytoponction ganglionnaire permet une mise en culture des cellules prélevées avec analyse bactériologique. Cet examen, moins invasif qu’une biopsie, a l’avantage d’être facile à réaliser. Cependant, comme pour tout examen cytologique, des faux négatifs sont possibles et une cytoponction normale ne permet pas d’éliminer formellement une localisation tumorale. En cas de forte suspicion clinique de tumeur, un examen histopathologique ganglionnaire obtenu par biopsie-exérèse chirurgicale ou par biopsie à l’aiguille est indispensable pour poser un diagnostic définitif.
B. Indications de biopsie
La biopsie (à l’aiguille) ou biopsie-exérèse (faite chirurgicalement) d’un ganglion est indiquée :
• s’il existe une forte suspicion de pathologie tumorale (sur la cytoponction par exemple) ;
• en cas d’adénopathie chronique (> 1 mois) :
– inexpliquée au terme du bilan initial chez l’adulte,
– éventuellement après un traitement d’épreuve par antibiotiques chez l’enfant en cas de syndrome inflammatoire associé.
En cas d’adénopathies multiples, le choix de l’adénopathie à biopsier peut être orienté par les données de la TEP-TDM (adénomégalie la plus hypermétabolique accessible à la biopsie).
C. Biopsie à l’aiguille
Du fait de son invasivité moindre, la biopsie à l’aiguille est privilégiée par rapport à la biopsie-exérèse chirurgicale. Celle-ci se réalise en ambulatoire, sous anesthésie locale, guidée par l’imagerie (échographie ou scanner). La biopsie à l’aiguille présente néanmoins des limites. Elle nécessite d’être réalisée par un préleveur expérimenté et avec une aiguille de calibre suffisant afin d’obtenir des prélèvements biopsiques de bonne taille. Par ailleurs elle est également limitée dans la représentativité de la lésion, induisant un risque de faux négatif. Ainsi, l’échantillonnage limité peut parfois être insuffisant pour faire le diagnostic anatomopathologique et la biopsie-exérèse peut être nécessaire.
D. Biopsie-exérèse ganglionnaire
Elle vise à prélever l’ensemble du ganglion repéré. Elle se réalise au bloc opératoire sous anesthésie locorégionale ou générale nécessitant une organisation plus importante qu’une biopsie à l’aiguille. Le prélèvement doit intéresser le ganglion le plus suspect en évitant si possible les territoires inguinaux car ceux-ci sont souvent le siège de remaniements fibreux compliquant l’analyse microscopique. Il est adressé à l’état frais (sans fixateur), au mieux protégé du dessèchement par une compresse imbibée de sérum physiologique ou du milieu de culture (ex : RPMI – Roswell Park Memorial Institute), sans délai, au laboratoire d’anatomie pathologique +++. Le pathologiste se charge de la gestion du prélèvement en fonction de sa taille et des hypothèses diagnostiques.
La suspicion d’un lymphome rend obligatoire :
• une congélation d’un fragment du ganglion et conservation à –80 °C (cryopréservation) pour d’éventuelles explorations moléculaires complémentaires ;
•
•
• une analyse cytogénétique (caryotype) après mise en culture ;
• une analyse en cytométrie en flux ;
• une analyse en bactériologie.
Cas particulier de l’examen extemporané :
Points clés
• La plupart des adénopathies sont d’origine infectieuse.
• L’examen anatomopathologique est un examen clé surtout pour le diagnostic des pathologies tumorales.
• Le prélèvement de choix est la biopsie-exérèse d’un ganglion mais en pratique la biopsie à l’aiguille est de plus en plus réalisée en 1re intention.
• La suspicion d’un lymphome rend obligatoire :
– une congélation d’un fragment du ganglion frais (sans fixateur) ;
– puis une fixation dans du formol tamponné pour examen morphologique, étude immunohistochimique et éventuellement FISH ;
• Le prélèvement doit donc être adressé à l’état frais (sans fixateur) sans délai au laboratoire d’anatomie pathologique.