
CHAPITRE VI – Pathologie thoracique – Pneumologie
Plan :
• Item 159 – Tuberculose de l’adulte et de l’enfant
• Item 211 – Sarcoïdose
• Item 309 – Tumeurs du poumon, primitives et secondaires
• Item 210 – Pneumopathie interstitielle diffuse
• Item 206 – Épanchement pleural liquidien
Item 159 – Tuberculose de l’adulte et de l’enfant
Auteure : Audrey Lupo
I. Introduction
II. Histoire naturelle de la tuberculose et lésions tissulaires
III. Diagnostic et prélèvements
Hiérarchisation des connaissances – Tableau 1
{kind=link}
I. Introduction
A. Définition de l’inflammation granulomateuse
• cellules épithélioïdes (correspondant à des macrophages ou histiocytes mononucléés ressemblant à une cellule épithéliale) ;
• cellules géantes multinucléées (issues de la fusion des cellules épithélioïdes) ;
• lymphocytes.
Cette lésion correspond au granulome épithélioïde et gigantocellulaire, également appelé granulome tuberculoïde. L’inflammation granulomateuse est parfois appelée « inflammation spécifique » car ses caractéristiques morphologiques sont évocatrices d’un agent causal. En revanche, même si elles sont fortement évocatrices de la tuberculose, elles ne sont pas pathognomoniques car elles peuvent se rencontrer dans de nombreuses autres circonstances, entre autres : la sarcoïdose, la maladie de Crohn ou certaines infections fongiques (cf. item 211).
B. Caractéristiques des mycobactéries
II. Histoire naturelle de la tuberculose et lésions tissulaires
La contamination se fait par transmission aérienne (interhumaine directe) par inhalation d’« aérosols » de particules infectieuses émises par le patient présentant une tuberculose, entraînant la primo-infection.
Plusieurs facteurs de risque sont reconnus comme favorisant cette infection : la dénutrition, la précarité en raison de la fréquente promiscuité, la provenance en zone d’endémie (Afrique, Asie du Sud-Est, Asie centrale, Amérique du Sud, Europe centrale et de l’Est), l’immunodépression (VIH, cancer, traitements immunosuppresseurs), le diabète et les âges extrêmes (< 5 ans et > 70 ans).
Il existe trois entités nosologiques de la maladie :
• primo-infection tuberculeuse (PIT) ;
• infection tuberculeuse latente (ITL) ;
• tuberculose maladie (TM).
A. Primo-infection tuberculeuse
Elle correspond à l’inoculation de Mycobacterium tuberculosis, transmis le plus souvent par voie aérienne.
B. Infection tuberculeuse latente
La PIT évolue vers une phase de latence, résultat de l’équilibre entre le système immunitaire du patient et les bactéries, dont la durée dépend du statut immunitaire du patient. L’infection latente est par définition asymptomatique et n’a donc pas de traduction clinique ou radiologique. Elle peut être diagnostiquée par deux tests immunologiques mettant en évidence la réaction immunitaire à médiation cellulaire : l’intradermoréaction (IDR) à la tuberculine et le test in vitro de détection de la production d’interféron gamma (QuantiFERON-TB® ou T-Spot.TB®). L’ITL évolue vers la TM chez environ 10 % des adultes immunocompétents, généralement dans les 2 ans qui suivent la PIT.
C. Tuberculose maladie
Elle correspond à la phase symptomatique de la maladie et est liée à une réactivation au cours d’une baisse de l’immunité (âge extrême, diabète, immunodépression, etc.). Elle peut atteindre différents organes mais la forme pulmonaire (lieu de la contamination) reste majoritaire (75 % chez l’adulte immunocompétent).
1. Localisation pulmonaire
Forme classique
La réaction inflammatoire granulomateuse avec nécrose caséeuse détruit le parenchyme pulmonaire.
{kind=link}
Miliaire tuberculeuse
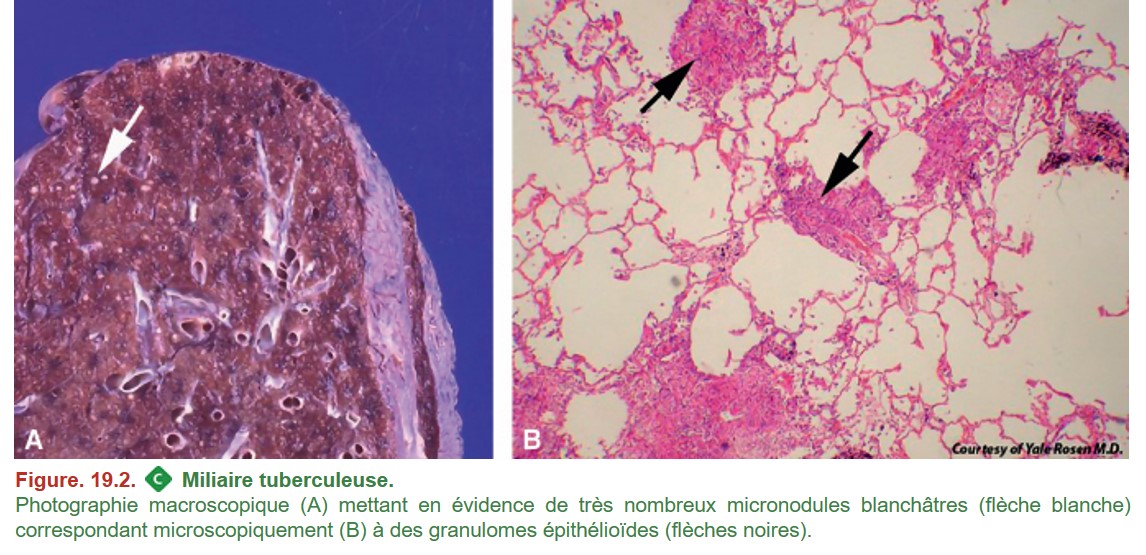{kind=link}
2. Localisations extrapulmonaires
Après dissémination hématogène, tous les organes peuvent être atteints. La localisation ganglionnaire, notamment cervicale, est la plus fréquente. Les autres localisations peuvent être : pleurale, péricardique, osseuse, notamment rachidienne avec une spondylodiscite (mal de Pott), neuroméningée, génito-urinaire, etc.
III. Diagnostic et prélèvements
Le diagnostic de tuberculose repose sur la mise en évidence du germe (Mycobacterium tuberculosis, rarement bovis ou africanum). Celle-ci est souvent difficile et nécessite des prélèvements respiratoires multiples et répétés : crachats, tubages gastriques, biopsies bronchiques, urines, liquide cérébrospinal, etc. Si des prélèvements tissulaires sont faits, ils doivent être adressés pour analyse bactériologique et aussi en anatomopathologie car les signes cliniques et radiologiques de la tuberculose peuvent faire évoquer de multiples pathologies pulmonaires et l’examen histologique permet d’éliminer un autre diagnostic.
A. Bactériologie des liquides et prélèvements multiples
•
• La culture sur milieux spéciaux solides (Löwenstein-Jensen) ou liquides, avec l’apparition des colonies pas avant 10-15 jours sur milieu liquide et 3-4 semaines sur milieu solide, permet l’identification du germe (sous-type de mycobactérie) et la réalisation d’un antibiogramme qui est obligatoire.
• Un test génotypique se fait pour tout examen direct ou culture positive. Il s’agit d’une amplification génique par PCR permettant de confirmer qu’il s’agit d’une mycobactérie du complexe tuberculosis et de rechercher la présence de mutation sur les gènes qui codent la résistance aux antituberculeux.
B. Anatomie pathologique
L’examen anatomopathologique n’est pas indispensable au diagnostic, il permet néanmoins d’orienter vers ce diagnostic ou d’éliminer un autre diagnostic que celui de la tuberculose. La lésion histologique élémentaire de la tuberculose est la présence de granulomes épithélioïdes et gigantocellulaires avec nécrose de type caséeuse centrale (figure 19.3) et la coloration de Ziehl-Neelsen permet de mettre en évidence les bacilles (BAAR) (figure. 19.4). Cette coloration est peu sensible et sa négativité n’élimine pas le diagnostic +++ car les bacilles sont généralement très peu nombreux dans les lésions granulomateuses et dans la nécrose caséeuse. De même, la nécrose caséeuse n’est pas présente dans toutes les lésions granulomateuses et son absence n’élimine pas le diagnostic.
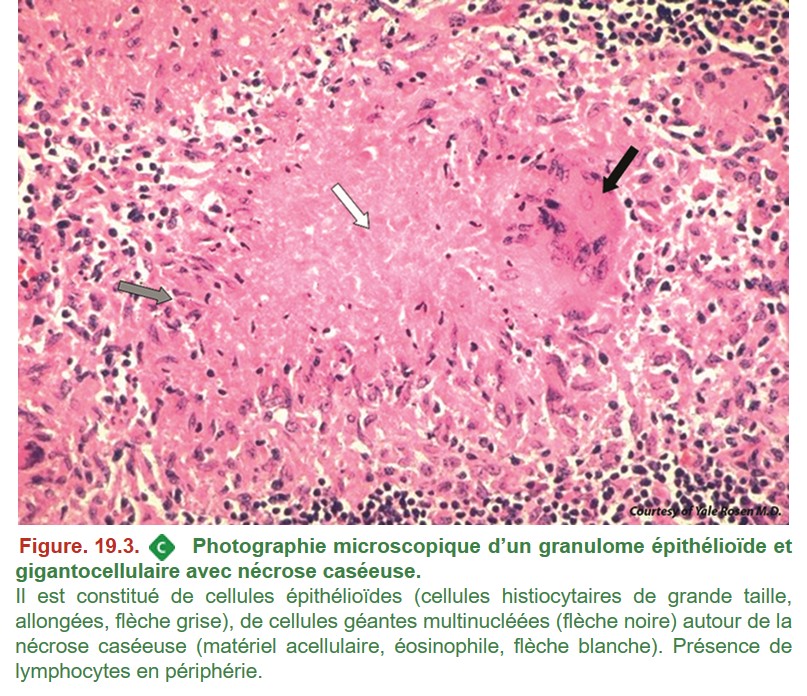{kind=link}
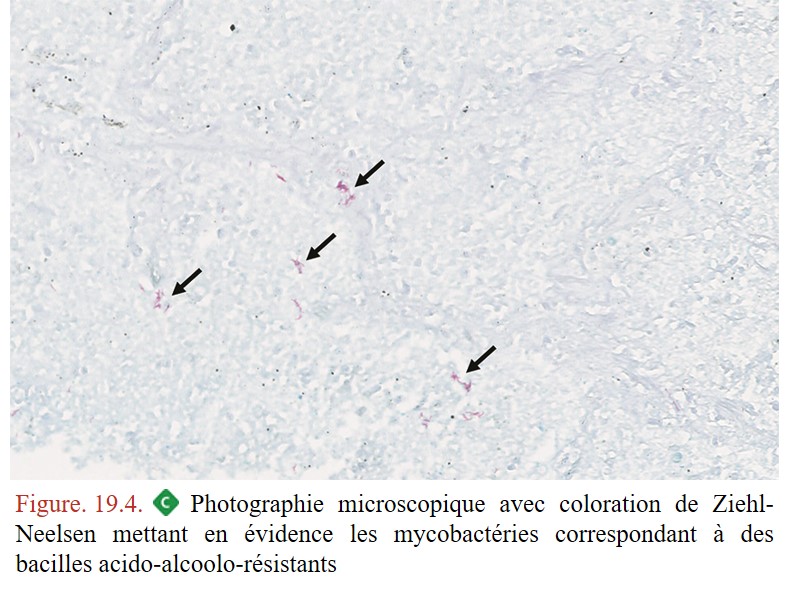{kind=link}
Définition :
Points clés
• Le diagnostic de tuberculose repose sur la mise en évidence des mycobactéries avec un envoi en bactériologie permettant l’identification de la mycobactérie et l’antibiogramme.
• En cas de prélèvements tissulaires, ces derniers doivent être adressés en anatomie pathologique en plus de la bactériologie, notamment pour éliminer un autre diagnostic.
• La lésion élémentaire tissulaire très en faveur du diagnostic est le granulome épithélioïde et gigantocellulaire avec nécrose caséeuse centrale.
• La recherche des BAAR visibles grâce à la coloration de Ziehl-Neelsen permet d’affirmer la présence de mycobactéries.
• L’absence de BAAR après coloration de Ziehl ou l’absence de nécrose caséeuse n’élimine pas le diagnostic (la nécrose caséeuse est inconstante et les BAAR sont peu nombreux dans les lésions granulomateuses).
Item 211 – Sarcoïdose
Auteure : Audrey Lupo
I. Prérequis
II. Introduction
III. Lésion élémentaire histologique
IV. Prélèvements pour mise en évidence des granulomes
V. Diagnostics différentiels anatomopathologiques
Hiérarchisation des connaissances : Tableau 2
{kind=link}
I. Prérequis
Définition de l’inflammation granulomateuse (cf. item 159).
II. Introduction
A. Définition de la sarcoïdose
B. Critères diagnostiques
Son diagnostic repose sur l’association de trois éléments :
• tableau clinique, biologique et radiologique évocateur ou compatible ;
• mise en évidence de granulomes épithélioïdes ± gigantocellulaires sans nécrose caséeuse (preuve histologique obligatoire sauf en cas de syndrome de Löfgren) ;
• élimination des autres causes de granulomatoses (en particulier la tuberculose).
Tous les organes peuvent être touchés mais les formes médiastino-pulmonaires (atteinte des poumons et des ganglions lymphatiques) sont les plus fréquentes (90 % des patients).
III. Lésion élémentaire histologique
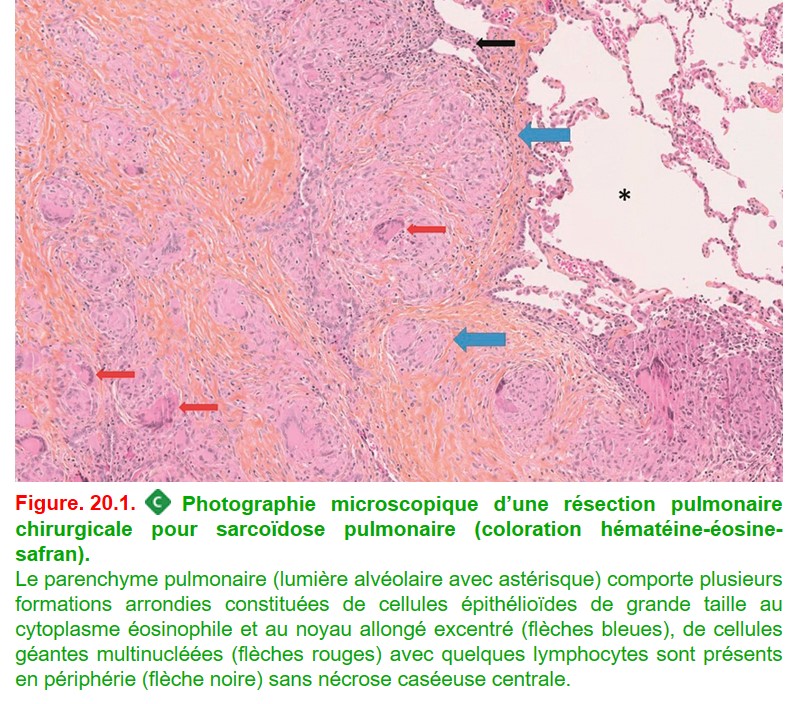
C’est le granulome épithélioïde et gigantocellulaire sans nécrose caséeuse (figure. 20.1).
{kind=link}
• des cellules épithélioïdes (cellules dérivant des phagocytes mononucléés et ressemblant à une cellule épithéliale avec une grande taille, un cytoplasme éosinophile et un noyau allongé excentré) ;
• des cellules géantes multinucléées (avec plusieurs noyaux) provenant de la fusion des cellules épithélioïdes ;
• un infiltrat lymphocytaire T CD4 + organisés en couronne autour des cellules épithélioïdes ;
• l’absence de nécrose caséeuse (à l’exception des rares formes de sarcoïdose nécrosante notamment chez le patient immunodéprimé).
Les granulomes épithélioïdes ± gigantocellulaires sont la conséquence d’une réaction immunitaire à médiation cellulaire (de type Th1), spécifique dirigée contre un antigène qui, dans la sarcoïdose, est encore inconnu (environnemental ou infectieux ?). Cette réaction survient généralement sur un terrain génétique prédisposant (plus fréquent chez les patients d’origine afro-antillaise). Au niveau pulmonaire, les granulomes sont souvent multiples, de petite taille, de distribution périlymphatique (dans le tissu conjonctif péribronchovasculaire et sous-pleural), responsables d’un syndrome interstitiel (cf. item 210). Le plus souvent, le granulome évolue spontanément ou sous traitement vers la résolution sans séquelle. Plus rarement, il persiste pendant plusieurs années sans altération de l’architecture de l’organe concerné ou évolue vers une fibrose.
IV. Prélèvements pour mise en évidence des granulomes
On privilégie dans un premier temps les sites d’accès aisé :
• biopsies des glandes salivaires accessoires :
– site le plus accessible,
– rentabilité moyenne (présence de granulomes chez 40 % des patients) ;
• biopsies de lésions cutanées :
– ne jamais biopsier un érythème noueux. Bien que la sarcoïdose soit la cause la plus fréquente d’érythème noueux en France, l’aspect histologique est aspécifique et ne contient jamais de granulome,
– les autres lésions cutanées (nodules, plaques ou lupus pernio) sont spécifiques et contiennent des granulomes ;
• biopsies d’une adénopathie superficielle :
– ces ganglions sont le plus souvent de petite taille, indolores et sans réaction inflammatoire cutanée périphérique,
– les localisations les plus courantes sont au niveau des aires ganglionnaires cervicales, sus-claviculaires et axillaires ;
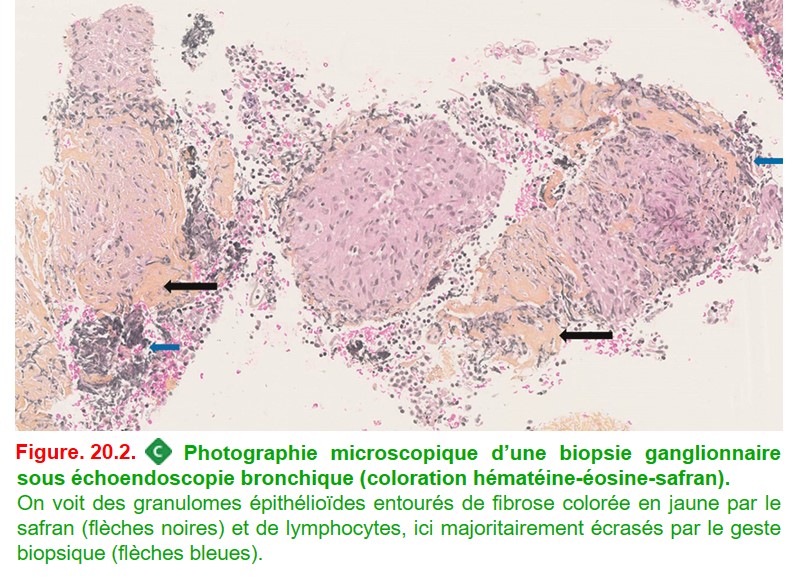
Puis, compte tenu de l’atteinte pulmonaire fréquente, on réalise des prélèvements au cours d’une endoscopie bronchique :
• biopsies bronchiques étagées proximales :
– au cours d’une fibroscopie bronchique avec lavage bronchoalvéolaire (LBA) à faire avant les biopsies,
– muqueuse d’aspect normal ou présentant des granulations jaunâtres, ou bien un épaississement des éperons (zones de bifurcation bronchique),
– sensibilité de 50 % seulement,
–
•
– technique : cf. item 210 ;
– hypercellularité ≈ 600 × 103 cellules/mL (pour une normale < 200 × 103 cellules/mL chez un non-fumeur),
– nombreux lymphocytes (> 20 % de lymphocytes dans le LBA),
– prédominance de lymphocytes T CD4+ avec ratio CD4/CD8 typiquement > 3,5 (valeur normale < 2).
•
{kind=link}
Plus rarement, on discute la réalisation de :
• biopsies transbronchiques étagées : en cas d’atteinte parenchymateuse. Elles sont plus sensibles que les biopsies bronchiques mais avec un risque hémorragique et de pneumothorax ;
• médiastinoscopie en cas d’atteinte ganglionnaire médiastinale, examen le plus sensible (90-100 %) lorsque l’EBUS n’a pas permis de diagnostic ;
• biopsies hépatiques : en cas de perturbation du bilan hépatique, la sensibilité est bonne (60 %)
Les autres sites atteints (œil, cœur, rate, etc.) ne sont classiquement pas biopsiés.
V. Diagnostics différentiels anatomopathologiques
Les principales autres causes de granulomes épithélioïdes sans nécrose caséeuse sont les suivantes :
• granulomatoses infectieuses ++ :
– tuberculose et autres mycobactéries : rechercher une nécrose au centre des granulomes et la présence de BAAR (coloration de Ziehl) (cf. item 159),
– mycose (histoplasmose ++) :
–
– infections à Chlamydia, Bartonella ou Yersinia pseudotuberculosis : présence de granulomes épithélioïdes parfois suppurés (centrés par une nécrose avec polynucléaires neutrophiles),
– instillation intravésicale thérapeutique de BCG (forme atténuée de Mycobacterium bovis) dans les tumeurs vésicales, BCGite (réaction cutanée sur site injection du vaccin), ;
• granulomes à corps étrangers ;
• maladies d’exposition (bérylliose ++, silicose) ;
• médicaments :
– granulomatoses hépatiques (isoniazide, quinine, diltiazem, carbamazépine, interféron, etc.) et rénales (bêtalactamines, rifampicine, AINS, paracétamol),
– granulomes épithélioïdes et gigantocellulaires développés chez les patients sous immunothérapie par anticorps anti-PD-1 ou PD-L1, de même aspect que ceux de la sarcoïdose : importance du contexte clinique ++,
– pneumopathie d’hypersensibilité médicamenteuse ;
• réactions granulomateuses au cours des cancers que l’on peut retrouver en périphérie des lésions tumorales (lymphome de Hodgkin, lymphome T, etc.) ou dans les ganglions des territoires de drainage des cancers, d’origine paranéoplasique. On peut également retrouver ces granulomes en réponse à un traitement par immunothérapie ;
• autres maladies inflammatoires auto-immunes : maladie de Crohn, cholangite biliaire primitive, etc.
• déficit immunitaire commun variable : groupe hétérogène de maladies caractérisées par une hypogammaglobulinémie avec déficit de production d’anticorps spécifiques après immunisation. Il provoque des infections bactériennes récidivantes, en général par bactéries encapsulées (pouvant donner des granulomes) ;
• restauration immunitaire par traitement anti-VIH.
• pneumopathies d’hypersensibilité : granulomes de petite taille, interstitiels ou péricentrolobulaires, vus sur les prélèvements de poumon distal ou biopsies transbronchiques, pas sur les biopsies bronchiques.
Points clés
• La sarcoïdose est une affection systémique, d’étiologie inconnue.
• L’atteinte médiastinopulmonaire est la manifestation clinique la plus fréquente (90 % des patients).
• Le diagnostic de sarcoïdose repose sur l’association de :
– la clinique et notamment la connaissance des pathologies associées et traitements reçus dans le cadre du diagnostic différentiel ;
– la mise en évidence de granulomes épithélioïdes ;
– l’élimination des autres causes d’inflammations granulomateuses (en particulier infectieuses, en premier lieu la tuberculose +++).
• La lésion élémentaire histologique est un granulome épithélioïde ± gigantocellulaire sans nécrose caséeuse.
• La stratégie diagnostique doit être adaptée à la recherche des granulomes pour être la moins invasive possible (biopsie de sites périphériques).
Item 309 – Tumeurs du poumon, primitives et secondaires
Auteure : Audrey Lupo
I. Prérequis
II. Tumeurs primitives du poumon
III. Tumeurs secondaires du poumon (métastases)
Hiérarchisation des connaissances – Tableau 3
{kind=link}
I. Prérequis
A. Anatomie
Le poumon gauche est formé de deux lobes et le poumon droit de trois lobes. Le médiastin est situé entre les deux poumons, il est délimité à l’avant par le sternum, à l’arrière par la colonne vertébrale, en haut par le cou et en bas par le diaphragme. La plèvre est formée de deux feuillets : le feuillet viscéral qui tapisse les lobes pulmonaires et le feuillet pariétal qui tapisse latéralement la paroi thoracique (côtes), en bas le diaphragme et en interne le médiastin.
B. Histologie
Poumon = arbre bronchique (proximal) + parenchyme périphérique (périphérique).
• Arbre bronchique : il est tapissé d’un épithélium respiratoire pseudostratifié constitué de cellules épithéliales ciliées ou mucosécrétantes et de quelques cellules épithéliales neuroendocrines. Sous l’épithélium, on retrouve le chorion, la musculeuse, puis la sous-muqueuse qui contient les glandes bronchiques et le cartilage (figure. 21.1).
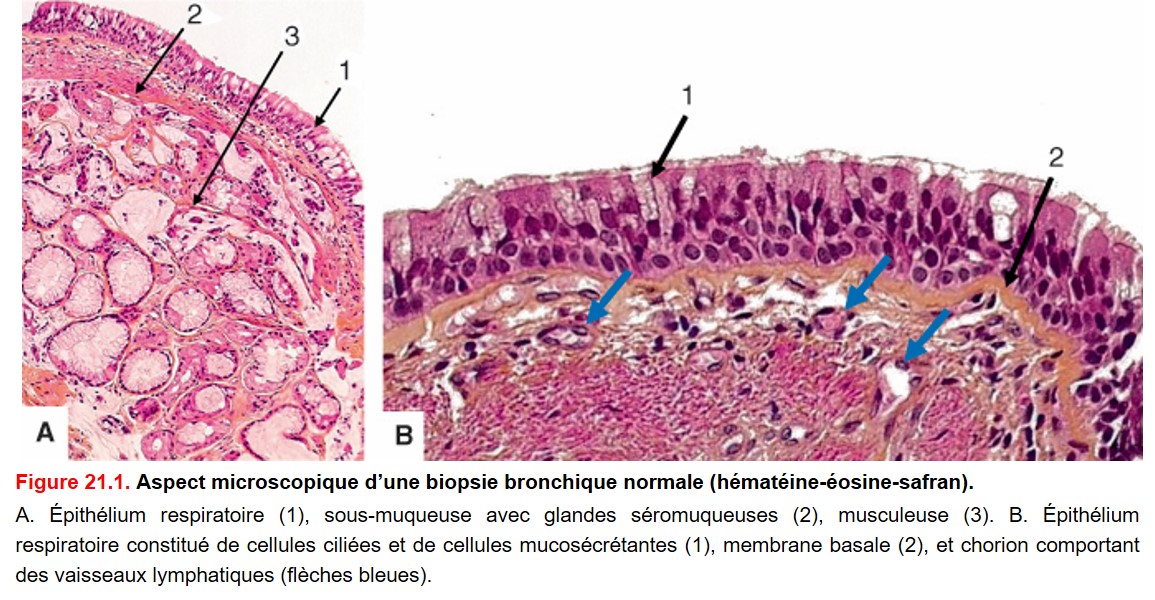{kind=link}
• Parenchyme périphérique : les bronches se divisent successivement pour donner les bronchioles terminales, puis les bronchioles respiratoires et enfin les alvéoles (figure. 21.2). Les alvéoles sont tapissées de pneumocytes (de type I qui participent aux échanges gazeux et de type II qui synthétise le surfactant, ces derniers expriment TTF-1 : thyroid transcription factor). Les lumières alvéolaires comportent des macrophages.
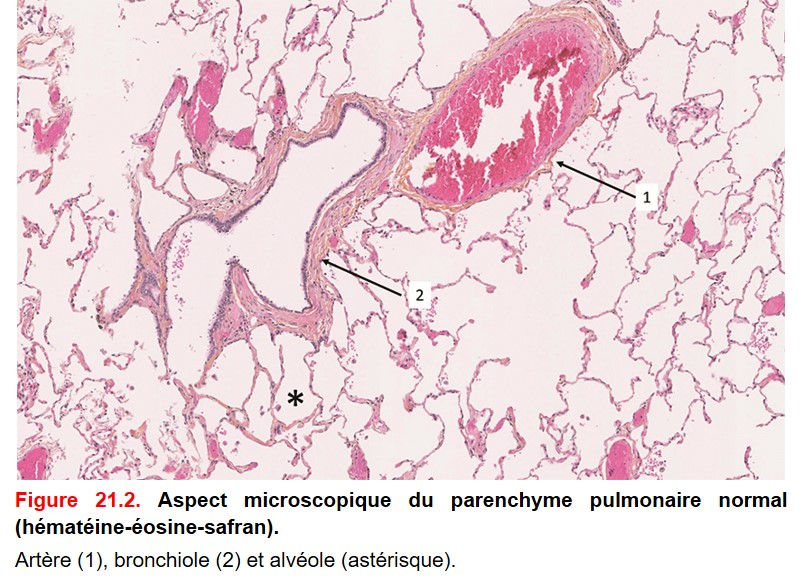{kind=link}
II. Tumeurs primitives du poumon
A. Épidémiologie
Plus de 3/4 des patients sont diagnostiqués à un stade localement avancé (stade III) ou métastatique (IV). L’espérance de vie à 5 ans tous stades confondus est inférieure à 20 %.
B. Facteurs de risque et oncogenèse
1. Tabac
C’est le facteur de risque principal, 85 % des cancers pulmonaires sont dus au tabac. Le risque carcinogène augmente avec la précocité d’âge de début du tabagisme et la durée du tabagisme. Après l’arrêt du tabac, le risque décroît mais ne revient jamais au niveau de celui du non-fumeur. L’exposition passive au tabac augmente le risque de cancer du poumon et on estime qu’il est responsable du quart des cancers du poumon chez les patients non-fumeurs.
2. Autres facteurs de risque
Ils sont présentés dans le tableau 21.1.
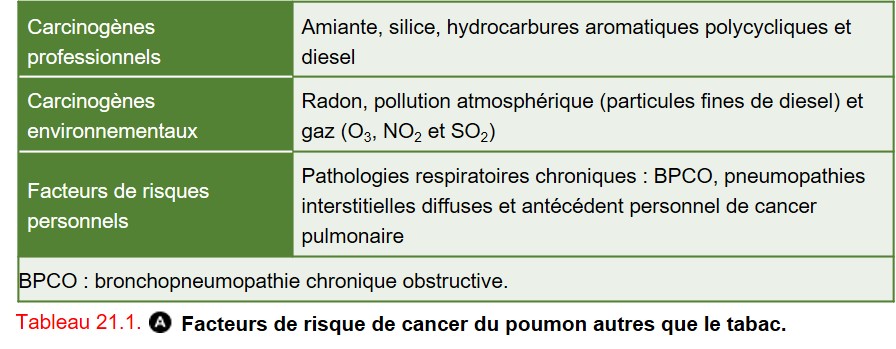{kind=link}
3. Oncogenèse
La carcinogenèse est différente selon l’origine tumorale.
Soit la tumeur est proximale, c’est-à-dire développée au niveau de l’arbre bronchique :
• métaplasie malpighienne de l’épithélium respiratoire ;
• dysplasie de l’épithélium malpighien (bas grade, haut grade/carcinome épidermoïde in situ) ;
• carcinome épidermoïde infiltrant.
Soit elle est développée en périphérie (parenchyme pulmonaire) :
•
•
•
La carcinogenèse évolue en plusieurs étapes, impliquant une série de mutations entraînant l’activation d’oncogènes. Dans certains cancers pulmonaires, la survie tumorale est dépendante de l’activation d’un seul oncogène (on parle d’« addiction oncogénique ») et le blocage de cette voie oncogénique par des thérapies dites ciblées est responsable de l’arrêt de la prolifération tumorale. Plusieurs addictions oncogéniques pour lesquelles des thérapies ciblées ont été développées sont disponibles dans les cancers du poumon, comme les mutations activatrices des gènes EGFR, ALK, ROS1, RET, BRAF, HER2, MET. Ces altérations moléculaires sont plus fréquemment retrouvées dans les adénocarcinomes et plus particulièrement chez les non-tabagiques.
C. Types histologiques
La très grande majorité des tumeurs pulmonaires sont des carcinomes (tumeurs malignes épithéliales). La détermination précise du type histologique est essentielle pour définir la prise en charge thérapeutique. On distingue les carcinomes bronchopulmonaires non à petites cellules (CBNPC) (85 %) des carcinomes à petites cellules (CPC) (15 %), en raison de la présentation clinique, du pronostic et de la prise en charge thérapeutique qui sont différentes entre ces deux types (encadré 21.1).
{kind=link}
Parmi les CBNPC, les deux types les plus fréquents sont les adénocarcinomes et les carcinomes épidermoïdes dont la prise en charge thérapeutique est différente, notamment la prescription des thérapies ciblées qui n’est généralement proposée que dans les adénocarcinomes en raison des addictions oncogéniques retrouvées dans les adénocarcinomes et non dans les carcinomes épidermoïdes (hormis quelques rares cas de carcinomes épidermoïdes chez des patients non tabagiques).
1. Adénocarcinome
•
• Type histologique le plus fréquent chez les non-fumeurs.
• Histologie : tumeur maligne épithéliale avec différenciation glandulaire. Cette différenciation peut être reconnue morphologiquement par la présence de glandes (figure. 21.3) et/ ou d’une mucosécrétion. D’un point de vue immunohistochimique, ces tumeurs expriment dans 80-90 % des cas TTF-1 sans expression de p40.
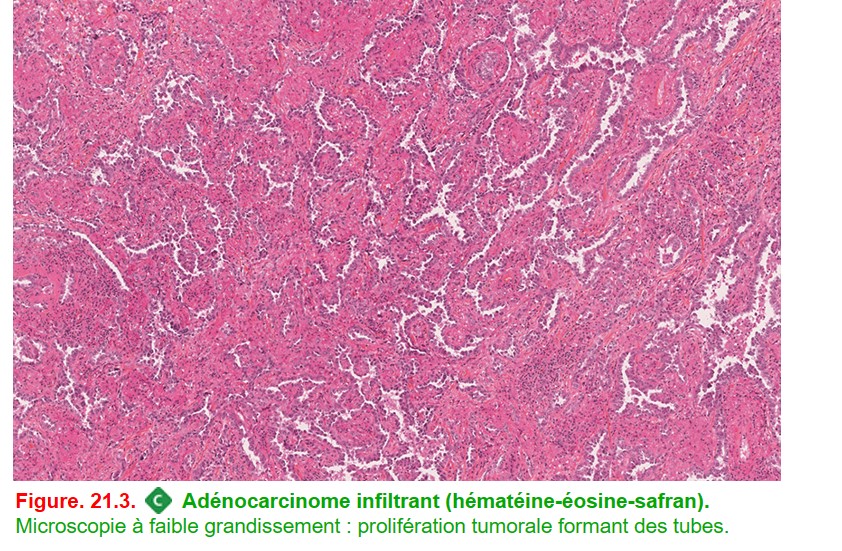{kind=link}
• Parfois associé à une addiction oncogénique permettant la prescription d’une thérapie ciblée
• Forme particulière : adénocarcinome in situ, de très bon pronostic (survie de 100 % à 5 ans chez les patients opérés), il s’agit d’une prolifération de cellules carcinomateuses le long des cloisons alvéolaires sans invasion stromale, vasculaire ou pleurale, ni métastases ganglionnaires ou à distance (figure. 21.4).
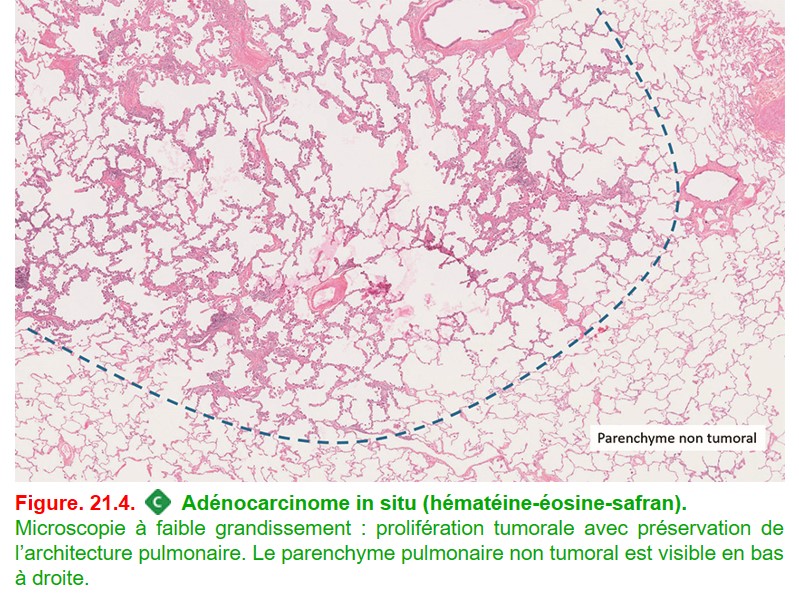{kind=link}
2. Carcinome épidermoïde
• Localisation le souvent proximale, péri ou endobronchique.
• Association forte avec le tabagisme.
• Histologie : tumeur maligne épithéliale avec différenciation malpighienne. Cette différenciation peut être reconnue morphologiquement par la présence de ponts d’union intercellulaires ou de kératine (figure. 21.5). D’un point de vue immunohistochimique, ces tumeurs expriment p40 sans expression de TTF-1.
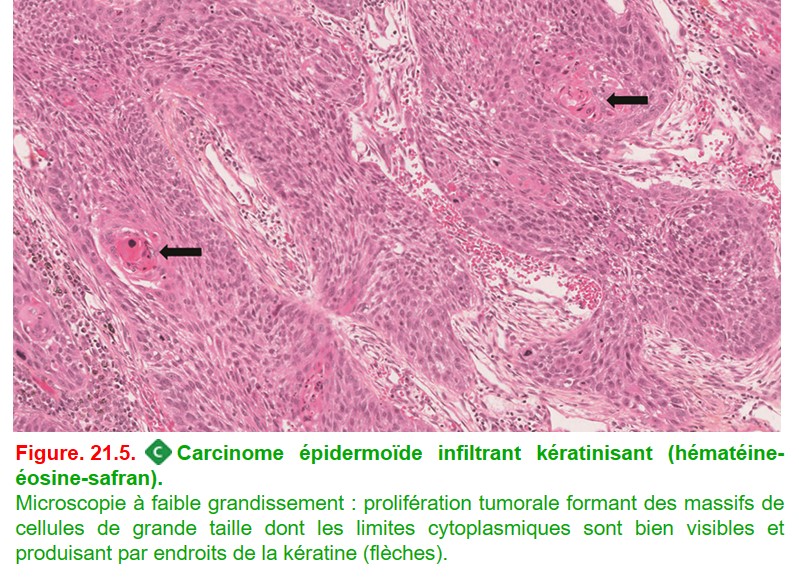{kind=link}
3. Carcinome à petites cellules
Les carcinomes à petites cellules font partie du groupe des tumeurs neuroendocrines qui comprend également les tumeurs carcinoïdes et les carcinomes neuroendocrines à grandes cellules.
•
• Association quasi constante avec un tabagisme important.
• Souvent associé à un syndrome paranéoplasique.
• Son pronostic est mauvais en raison des rechutes et de l’acquisition rapide de chimiorésistance.
•
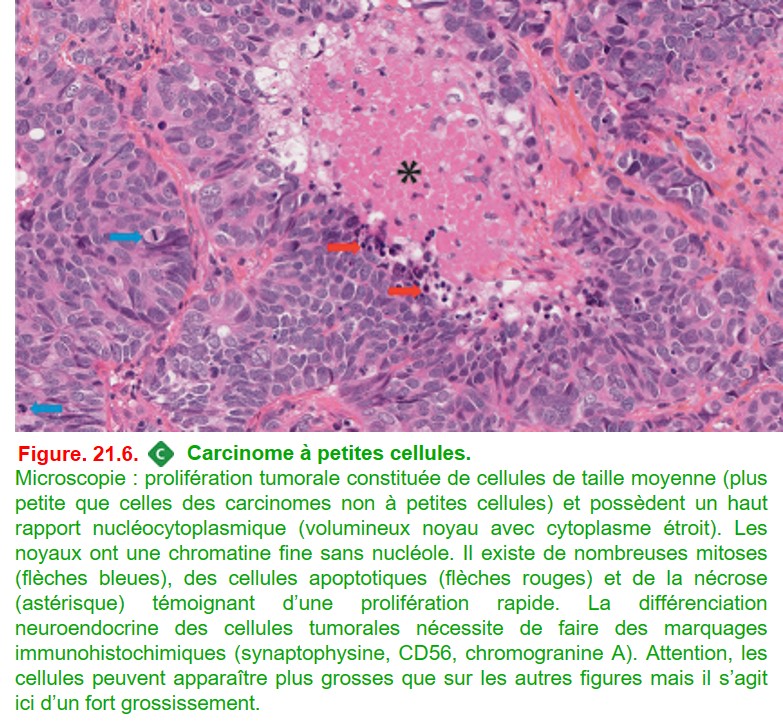{kind=link}
D. Démarche diagnostique et prélèvements
Les prélèvements réalisés dans le cadre d’un diagnostic de cancer pulmonaire dépendent de l’extension de la maladie, qui dans plus de 75 % des cas est de stade localement avancé ou métastatique. On peut prélever la tumeur pulmonaire ou la localisation métastatique (ganglionnaire ou viscérale) de la tumeur.
La démarche diagnostique inclut le prélèvement histologique et le bilan d’extension permettant de réaliser le stade TNM qui est un des facteurs pronostiques les plus importants. Le stade TNM permet d’évaluer l’extension de la tumeur aux structures locorégionales (T), aux ganglions (N : node = ganglion en anglais) et aux organes à distance (M : métastase).
•
• Les stades III sont des stades localement avancés plutôt traités par radiochimiothérapie.
• Les stades IV métastatiques nécessitent un traitement systémique (chimiothérapie, immunothérapie, thérapie ciblée).
1. Méthodes de prélèvement
Endoscopie bronchique
D’autres prélèvements à visée diagnostique peuvent être faits pendant la fibroscopie :
• le brossage bronchique (analyse cytologique) ;
• le recueil de l’aspiration bronchique (analyse cytologique) ;
• le LBA (analyse cytologique) ;
• les ponctions échoguidées par voie endoscopique permettant de prélever des ganglions, à l’aide d’une sonde échographique située à l’extrémité de l’endoscope (EBUS).
Biopsie transpariétale
En cas de tumeur non accessible par voie endobronchique, une biopsie transpariétale peut être réalisée sous repérage scanographique, sous anesthésie locale.
Prélèvements chirurgicaux. Ils incluent :
• la médiastinoscopie permettant de prélever les ganglions du médiastin moyen (latérotrachéaux, sous-carénaire), à la recherche de métastases ganglionnaires ;
• la vidéothoracoscopie exploratrice avec biopsie chirurgicale pulmonaire qui est exceptionnellement employée, uniquement dans les cas où la biopsie transpariétale n’a pas permis un diagnostic ;
• l’exérèse d’une métastase (ex : surrénalectomie).
Prélèvements d’une lésion à distance
Ils ont pour objectif la biopsie d’une adénopathie, d’une métastase sous-cutanée, osseuse ou hépatique, voire d’une lésion cérébrale.
2. Choix de la technique de prélèvement
En règle générale, on privilégie les prélèvements peu invasifs plutôt que les prélèvements chirurgicaux.
• Le choix de la technique dépend également de la localisation de la tumeur pulmonaire (proximale ou périphérique).
• Les tumeurs proximales sont habituellement accessibles par fibroscopie bronchique.
• Les tumeurs périphériques sont prélevées généralement par ponction transpariétale à l’aiguille sous contrôle scanographique.
• Lorsqu’il existe une suspicion de localisation ganglionnaire médiastinale métastatique, on peut la documenter par un EBUS ou dans un second temps par une technique chirurgicale plus invasive (médiastinoscopie) si l’EBUS est négatif.
• Enfin, si la tumeur est disséminée, on peut prélever un des sites métastatiques le plus facilement accessible comme les métastases sous-cutanées, surrénaliennes, hépatiques ou osseuses.
L’avantage de prélever une lésion à distance est qu’elle permet, en plus du diagnostic, d’affirmer le stade métastatique de la maladie.
E. Stratégie thérapeutique
Les décisions thérapeutiques doivent être validées en RCP.
La prise en charge thérapeutique nécessite :
• une confirmation histologique et la précision du type histologique ;
• un bilan d’extension permettant de déterminer le stade de la maladie (TNM) ;
• un bilan préthérapeutique (état général du patient, bilan nutritionnel, bilan cardiovasculaire, fonction respiratoire) permettant de décider des traitements qui pourraient éventuellement être contre-indiqués.
1. Carcinomes non à petites cellules
• Stade localisé (I et II) : il ne représente qu’un tiers des patients, la chirurgie est la meilleure option thérapeutique si le patient est opérable, plus ou moins associée à une chimiothérapie.
•
• Stade métastatique (IV) : environ 50 % des patients.
2. Carcinomes à petites cellules
•
• Stade métastatique : chimiothérapie à base de sels de platine combinée à l’immunothérapie.
III. Tumeurs secondaires du poumon (métastases)
Le poumon est un site métastatique privilégié pour de nombreuses tumeurs malignes, comme les carcinomes extrapulmonaires ou pulmonaires, les mélanomes ou les sarcomes, et les métastases pulmonaires peuvent être révélatrices du cancer.
Selon l’incidence de ces tumeurs, les sites primitifs les plus probables devant une métastase pulmonaire sont dans l’ordre : sein, côlon, pancréas, estomac, peau, rein, ovaire. Elles peuvent se présenter sous forme d’un nodule pulmonaire unique (mélanome, rein, ovaire, thyroïde, etc.), ou de nodules pulmonaires multiples prenant un aspect en « lâcher de ballons » (sarcome, sein, testicule, etc.), ou de lymphangite carcinomateuse (sein, estomac, prostate, etc.).
L’étude anatomopathologique a donc pour but :
• de préciser le type et sous-type histologique de cancer (mélanome, sarcome, carcinome épidermoïde, adénocarcinome, carcinome neuroendocrine, etc.) ;
• en cas d’adénocarcinome, d’orienter vers certains primitifs en fonction de l’expression de certains marqueurs immunohistochimiques;
• de rechercher des cibles thérapeutiques.
Points clés
Tumeurs primitives
• Le diagnostic de cancer du poumon nécessite une preuve histologique (obligatoire ++).
• Pour les tumeurs proximales, le diagnostic est le plus souvent obtenu par biopsie sous fibroscopie bronchique alors que pour les tumeurs périphériques, il est le plus souvent obtenu par biopsie transpariétale.
• Deux grands groupes de carcinomes pulmonaires (de pronostic et traitement différents) :
– les CBNPC (adénocarcinomes > carcinomes épidermoïdes, etc.) (85 %) ;
– les CPC (15 %).
• Des analyses moléculaires à visée théranostique (mutations EGFR, réarrangements de ALK et ROS1, etc.) doivent être effectuées systématiquement dans les CBNPC non épidermoïdes, de stade métastatique, ces analyses sont réalisées à partir de tissu inclus en paraffine.
• La recherche par immunohistochimie de l’expression de PD-L1 par les cellules tumorales doit être systématique pour tous les CBNPC de stade avancé ou métastatique. Le niveau d’expression de PD-L1 conditionne la prescription d’une immunothérapie seule ou combinée à la chimiothérapie par anticorps monoclonal dirigé contre PD-1 ou PD-L1, en absence d’altération moléculaire ciblable.
• Les CPC sont des tumeurs neuroendocrines avec positivité des marqueurs (synaptophysine, chromogranine A, CD56, etc.), ils sont de mauvais pronostic, relevant rarement d’un traitement chirurgical.
Tumeurs secondaires du poumon (métastases)
• Les métastases pulmonaires peuvent être révélatrices du cancer.
• L’étude anatomopathologique précise le type histologique de cancer, peut orienter vers la tumeur d’origine et permet de rechercher des cibles thérapeutiques.
Item 210 – Pneumopathie interstitielle diffuse
Auteure : Audrey Lupo, Aurélie Cazes
I. Généralités
II. Prélèvements cytologiques et histologiques diagnostiques
III. Aspects cytologiques et histologiques
Hiérarchisation des connaissances – Tableau 4
{kind=link}
I. Généralités
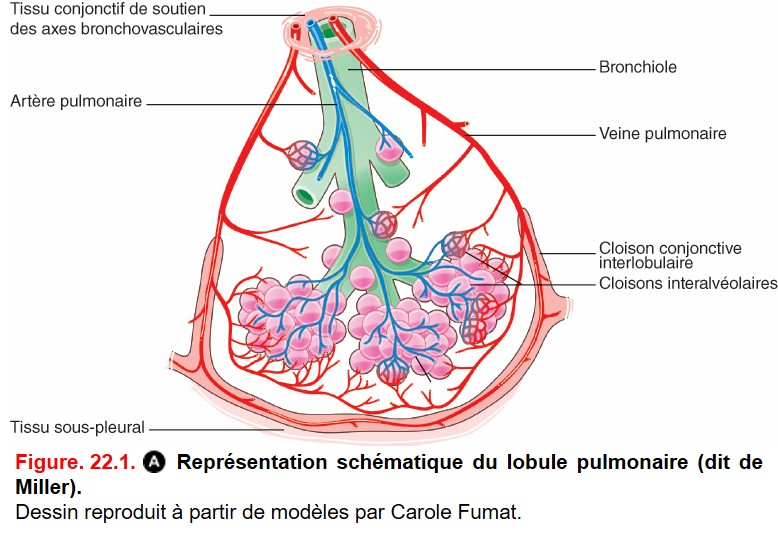
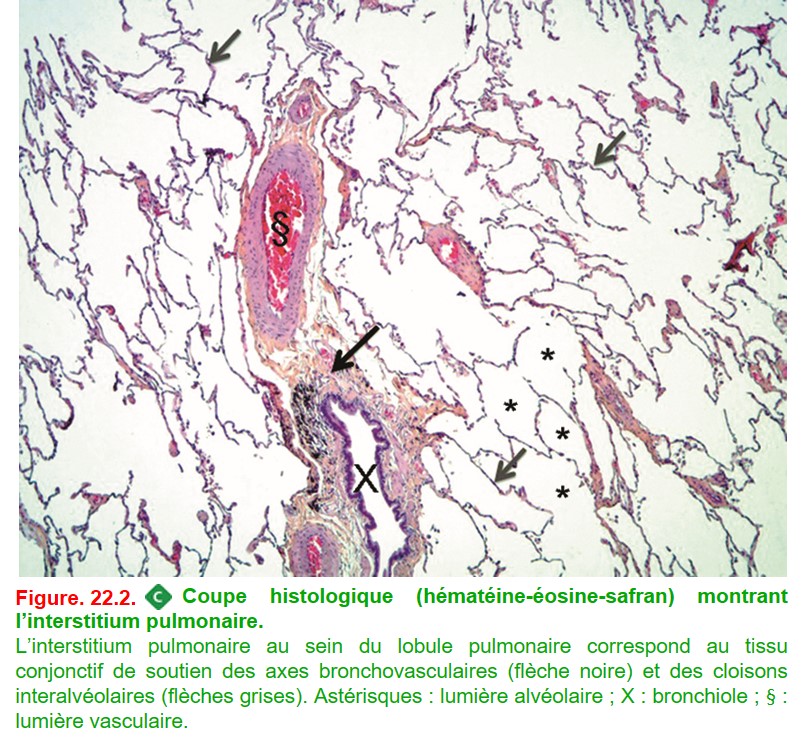
Anatomiquement, elles se caractérisent par une atteinte prédominante de l’interstitium pulmonaire (figure. 22.1 et figure 22.2), regroupant :
{kind=link}
{kind=link}
• le tissu conjonctif de soutien des axes bronchovasculaires ;
• les cloisons interlobulaires (du lobule secondaire de Miller) ;
• les cloisons interalvéolaires ;
• le tissu sous-pleural.
La topographie et l’atteinte de ces différentes structures histologiques sont différentes en fonction des pathologies. Par exemple :
• les lésions de la sarcoïdose sont majoritairement localisées autour des axes bronchiques ;
• les atteintes de la fibrose pulmonaire idiopathique sont majoritairement localisées au niveau de la cloison interlobulaire.
La démarche diagnostique est différente pour les PID aiguës et pour les PID chroniques.
Le diagnostic de PID est multidisciplinaire et repose sur un faisceau d’arguments : interrogatoire + présentation clinique + anomalies radiologiques pulmonaires (scanner +++) + explorations fonctionnelles respiratoires + lavage bronchoalvéolaire (cytologie) + examens biologiques ± prélèvement histologique.
II. Prélèvements cytologiques et histologiques diagnostiques
A. Lavage bronchoalvéolaire
Le LBA peut faire l’objet :
• d’une analyse microbiologique (obligatoire en cas de suspicion d’une pathologie infectieuse) ;
• d’une analyse cytologique ;
• d’autres analyses : recherche de corps asbestosiques ou de silice au LAFP (laboratoire amiante, fibres et particules ex-LEPI [laboratoire d’étude des particules inhalées]), immunophénotypage lymphocytaire (rapport CD4/CD8).
1. Aspects techniques
Au laboratoire d’anatomie et cytologie pathologiques, il est procédé à :
• la mesure du volume ;
• la description de l’aspect (couleur, viscosité, etc.) ;
• une numération (ou cellularité : nombre de cellules/ml) ;
• puis une cytocentrifugation du liquide permettra de former une petite pastille avec les cellules du LBA sur une lame (figure. 22.3) ;
{kind=link}
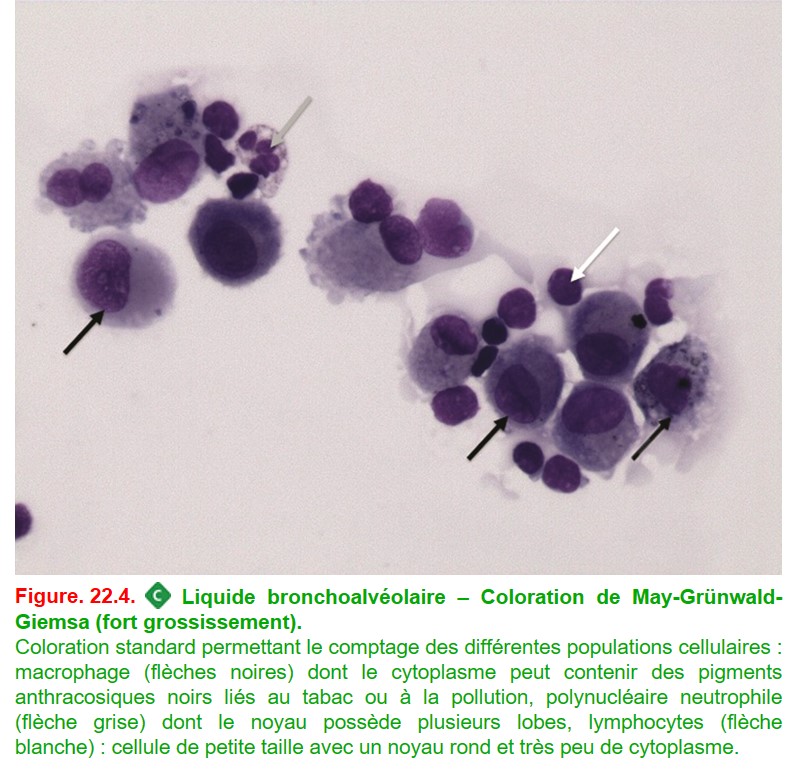
• une coloration systématique avec les colorations de :
– May-Grünwald-Giemsa (figure. 22.4) (distinction des cellules),
{kind=link}
– Papanicolaou (distinction des cellules, inclusions nucléaires virales, mise en évidence des cellules malpighiennes contaminantes),
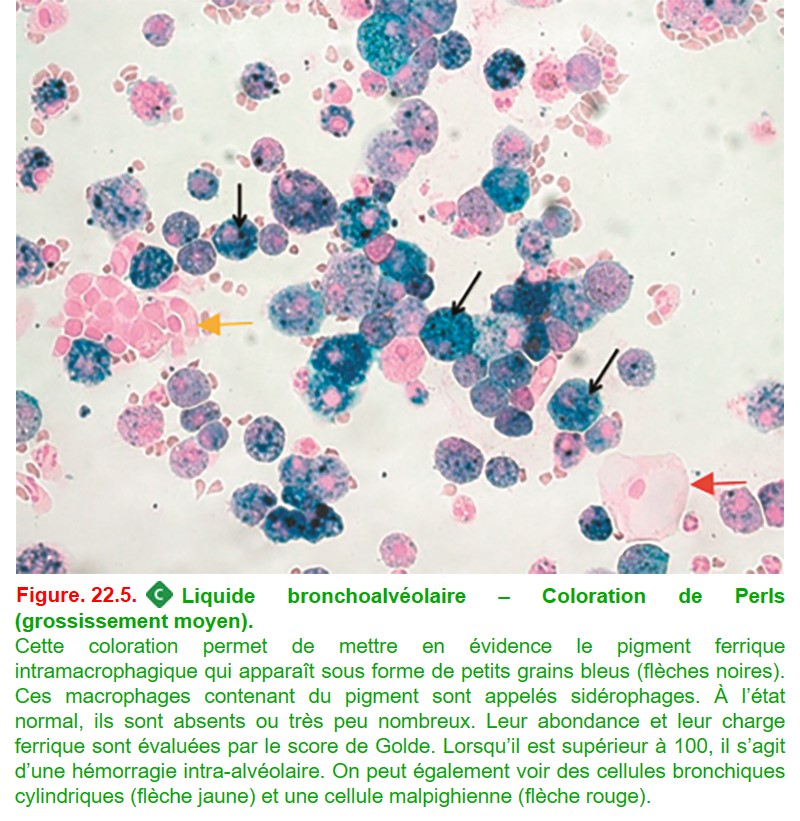
– Perls pour la recherche de sidérophages (fer) (figure. 22.5) ;
{kind=link}
• la conservation d’autres lames pour effectuer éventuellement d’autres colorations, notamment :
– à la recherche d’agents infectieux :
– Gomori-Grocott pour recherche de champignons,
– Gram pour la recherche de bactéries,
– Ziehl pour la recherche de mycobactéries ;
– ou pour d’éventuels marquages immunocytochimiques (ex : CD4, CD8 dans le cadre d’une suspicion de sarcoïdose, ou CD1a pour une histiocytose langerhansienne).
2. Analyse
Elle comprend, en plus de la mesure du volume et de la description de son aspect :
• l’établissement de la formule (répartition en pourcentage des différents types de cellules) ;
• une recherche d’éléments cellulaires anormaux (cellules tumorales, effet cytopathogène viral) ;
• une recherche d’agents pathogènes sur les colorations habituelles ou spéciales (champignons, parasites, mycobactéries, bactéries intracellulaires, etc.) ;
• une recherche de sidérophages sur la coloration de Perls (macrophages contenant du pigment hémosidérinique ou surcharge en fer témoignant d’une phagocytose d’hématies) avec établissement du score de Golde;
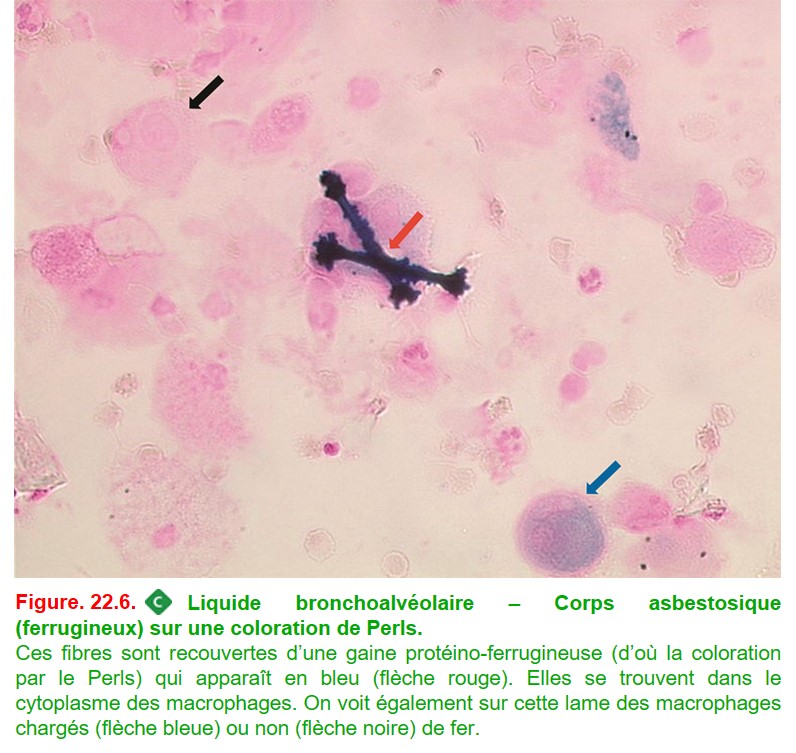
• une recherche de corps ferrugineux en faveur d’une exposition à l’amiante (figure. 22.6).
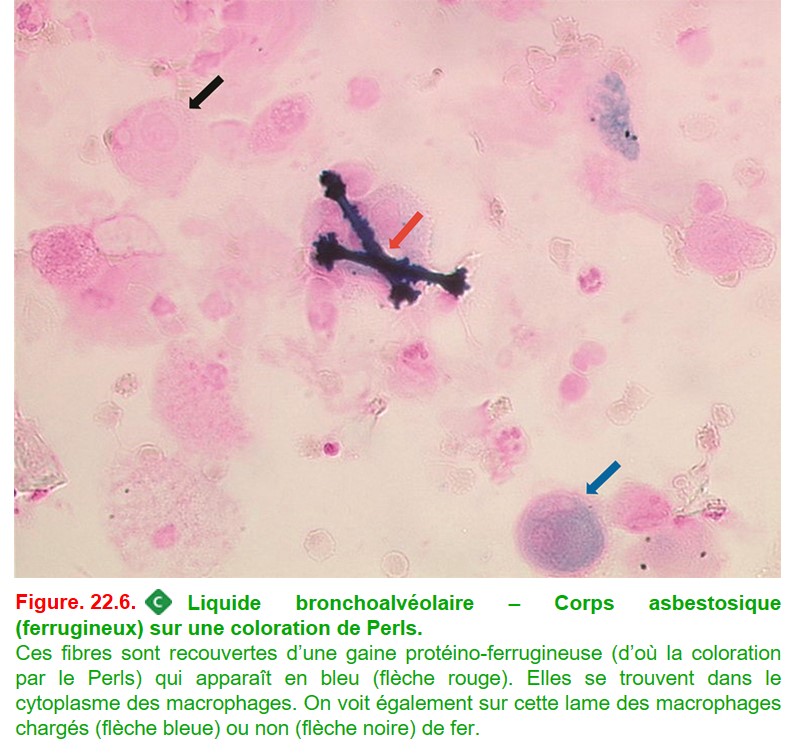{kind=link}
3. Aspect normal
• Aspect : clair.
• Cellularité : < 150 000 à 200 000 cellules/ml (sujet non fumeur).
• Composition cellulaire (formule) :
– macrophages : > 85 %,
– lymphocytes : 10-15 %, rapport CD4/CD8 normal : 1,5,
– polynucléaires neutrophiles : < 3 %,
– polynucléaires éosinophiles : < 1 %.
Les cellules bronchiques doivent rester très minoritaires, sinon il s’agit d’une contamination bronchique rendant le prélèvement non représentatif de l’alvéole.
4. Orientation diagnostique
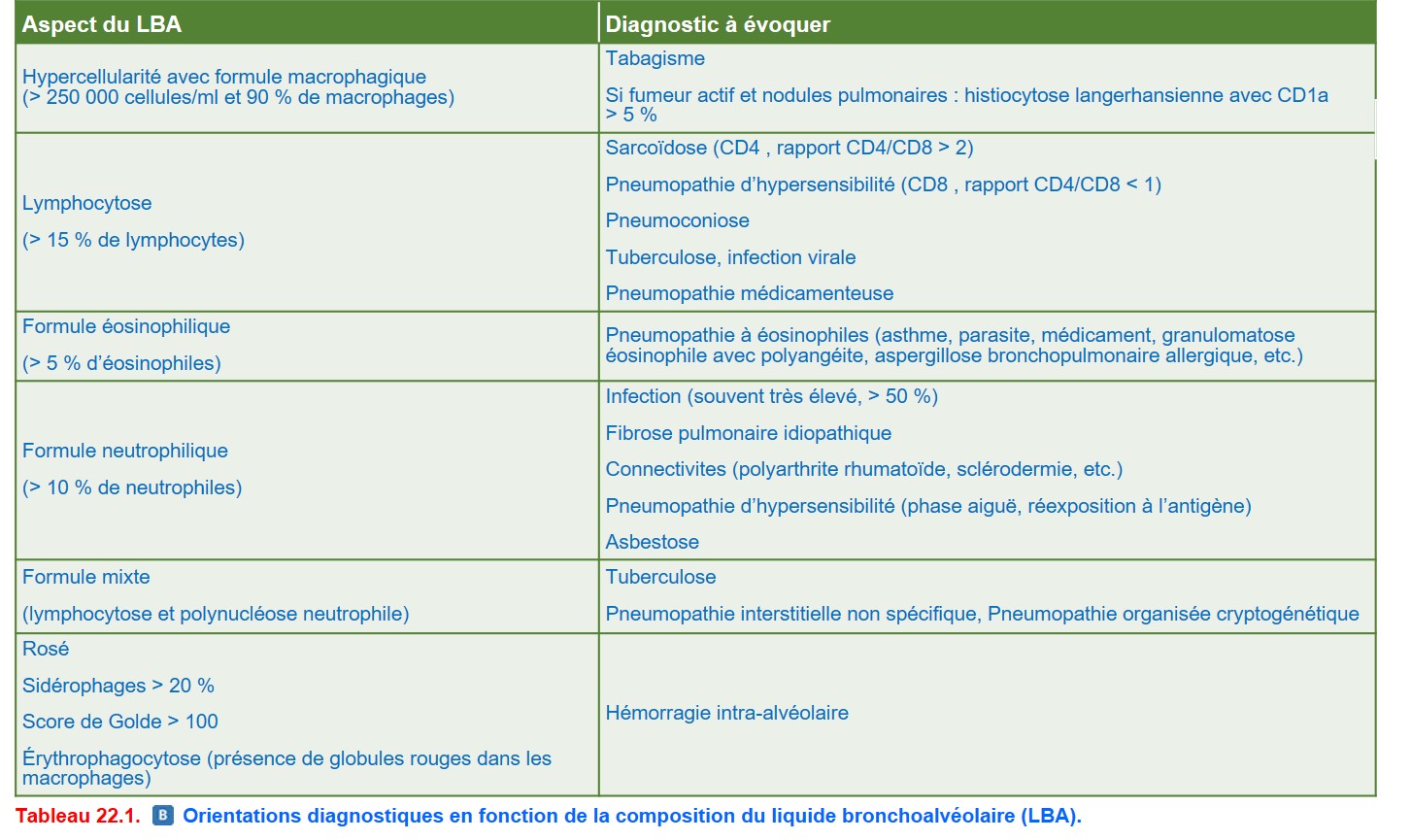{kind=link}
• la mise en évidence de cellules tumorales ;
• la mise en évidence de mycobactéries (coloration de Ziehl) ;
• la mise en évidence de matériel spumeux avec la présence de germes colorés par le Grocott correspondant à Pneumocystis jirovecii (figure. 22.7) ;
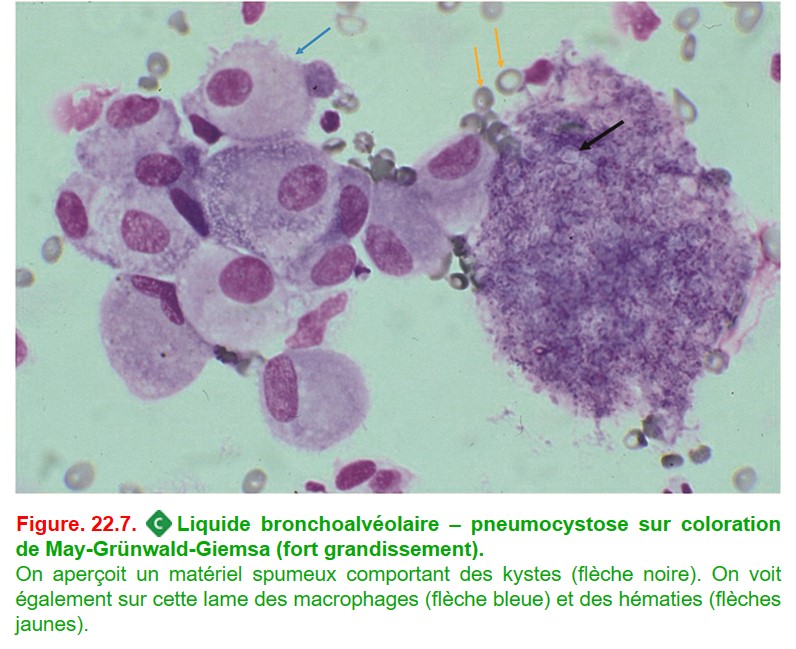{kind=link}
• le taux de sidérophages > 20-30 % pour l’hémorragie intra-alvéolaire, le score de Golde > 100.
B. Biopsies sous endoscopie bronchique
• Les biopsies bronchiques d’éperons ont une bonne rentabilité (> 50 % de sensibilité) pour les diagnostics de sarcoïdose ou lymphangite carcinomateuse dont les lésions s’observent de façon diffuse au niveau de la muqueuse et des lymphatiques
• Les biopsies pulmonaires transbronchiques (qui prélèvent le tissu péribronchique, des bronchioles terminales et quelques alvéoles adjacentes) ont également une bonne rentabilité dans ces deux dernières entités, mais comportent des risques de pneumothorax et d’hémoptysie et ne sont pas indiquées dans les PID fibrosantes atteignant le poumon distal
Remarque : Les biopsies radioguidées sous scanner sont plus invasives et ne rapportent pas plus de matériel. Elles jouent un rôle clé dans le diagnostic des lésions tumorales périphériques, mais ne sont pas indiquées dans le contexte de pneumopathie interstitielle diffuse.
C. Biopsies pulmonaires chirurgicales par vidéothoracoscopie
Lorsque les premières étapes diagnostiques cliniques et paracliniques, avec notamment l’analyse du scanner, n’ont pas permis de porter un diagnostic, il peut être nécessaire d’analyser le poumon sur des biopsies pulmonaires chirurgicales sous vidéothoracoscopie. Si possible, elles doivent être multiples et intéresser plusieurs lobes, en fonction des atteintes radiologiques.
D. Prélèvements extrapulmonaires
Ils dépendent des hypothèses diagnostiques :
• cytoponction ganglionnaire sous échoendoscopie de ganglions médiastinaux devant une (fort agrandissement) suspicion de sarcoïdose ;
• biopsies des glandes salivaires accessoires devant une suspicion de sarcoïdose ou de connectivite (syndrome de Goujerot-Sjögren).
Dans tous les cas, il est indispensable de fournir au pathologiste les éléments cliniques et radiologiques ainsi que les hypothèses diagnostiques.
III. Aspects cytologiques et histologiques
Les PID peuvent être classées en fonction de leur caractère aigu ou chronique et de leur cause, connue ou inconnue.
A. PID aiguës
Les PID aiguës (< 3 semaines) sont dominées par les causes infectieuses et cardiogéniques (œdème aigu du poumon) (tableau 22.2).
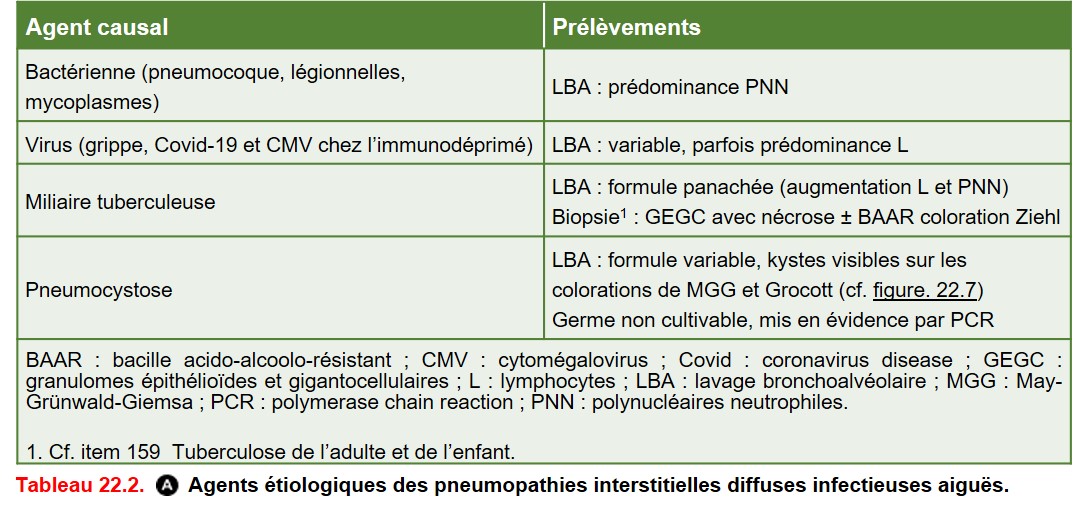{kind=link}
• Le LBA avec examen microbiologique et anatomopathologique est un examen important au cours des PID aiguës fébriles graves et chez l’immunodéprimé.
• Le bilan cardiologique est indispensable au cours des PID aiguës non fébriles et le LBA est rarement réalisé. S’il l’est, on observe un liquide hémorragique avec des polynucléaires neutrophiles et des pneumocytes altérés.
B. PID subaiguës ou chroniques
Il s’agit d’un groupe hétérogène d’affection que l’on peut séparer en PID de cause connue et de cause inconnue (encadré 22.1).
{kind=link}
En plus de la clinique (antécédents, interrogatoire) et des EFR (explorations fonctionnelles respiratoires), le diagnostic repose essentiellement sur :
• le scanner +++ ;
• la fibroscopie bronchique avec LBA ;
• des biopsies bronchiques (± transbronchiques) en cas de suspicion de sarcoïdose ou de lymphangite carcinomateuse.
1. Sarcoïdose
2. Pneumopathies infiltrantes diffuses idiopathiques
L’aspect du scanner est capital pour orienter le diagnostic +++.
Fibrose pulmonaire idiopathique (FPI)
• lésions d’âges différents (évolution par poussées) ;
• de répartition hétérogène et prédominant sous la plèvre ;
• avec des foyers fibroblastiques (lésions jeunes cellulaires) et de la fibrose constituée (lésions fixées, riches en collagène) ;
• résultant en une désorganisation et destruction architecturale avec aspect de rayon de miel.
Pneumopathie interstitielle non spécifique (PINS)
Pneumonie organisée idiopathique
Il s’agit d’une affection débutant sur un mode subaigu, dont l’aspect scanographique est évocateur. Le LBA n’est pas spécifique.
À la biopsie, l’architecture est préservée sans fibrose interstitielle. Des nodules conjonctifs (tissu de granulation) endoluminaux alvéolaires ou bronchiolaires sont présents.Ces lésions de pneumonie organisée peuvent également être de causes multiples (bactérienne, connectivite, radiothérapie, etc.).
3. Pneumoconioses
• L’asbestose est la plus fréquente des pneumoconioses, elle désigne la fibrose pulmonaire secondaire à l’inhalation de fibres d’amiante. Le LBA peut orienter le diagnostic en mettant en évidence des corps asbestosiques intramacrophagiques (cf. figure. 22.6). Ceci permet d’affirmer l’exposition et ne traduit pas forcément la maladie +++. L’atteinte au scanner prédomine en sous-pleural et aux bases, et des plaques pleurales calcifiées peuvent être associées.
{kind=link}
• La silicose est une pneumoconiose secondaire à l’inhalation de silice libre cristalline (tailleur de pierre ou d’ardoise). La biopsie pulmonaire montre des nodules dans l’interstitium bien limités de fibrose hyaline contenant des particules de silice biréfringentes en lumière polarisée et entourée d’histiocytes. L’atteinte au scanner prédomine dans les parties supérieures et des adénopathies en coquille d’œuf peuvent être associées.
L’interrogatoire doit être minutieux à la recherche d’une exposition. Il s’agit la plupart du temps de maladie professionnelle justifiant d’une reconnaissance par la sécurité sociale, l’exposition peut aussi être domestique. Une analyse minéralogique détermine avec précision le type de particule (prélèvement envoyé au LAFP).
4. Pneumopathie d’hypersensibilité
Les pneumopathies d’hypersensibilité (PHS) sont des affections liées à l’inhalation aiguë ou chronique d’antigènes. Les formes classiques historiques sont représentées par le « poumon d’éleveur d’oiseaux », principalement lié à l’exposition aux déjections, et le « poumon de fermier », principalement lié à l’exposition au foin moisi. La recherche d’une exposition aux poussières organiques, voire inorganiques à l’interrogatoire est donc primordiale. La détection de précipitines sériques (IgG) dirigées contre l’antigène inhalé permet de confirmer l’exposition. Le LBA est souvent lymphocytaire (> 50 %), notamment CD8+ (rapport CD4/CD8 < 1). Le scanner thoracique est souvent évocateur. Au scanner comme en histopathologie, on distingue les PHS non fibrosantes des PHS fibrosantes.
5. Origine tumorale
La présentation est plutôt subaiguë.
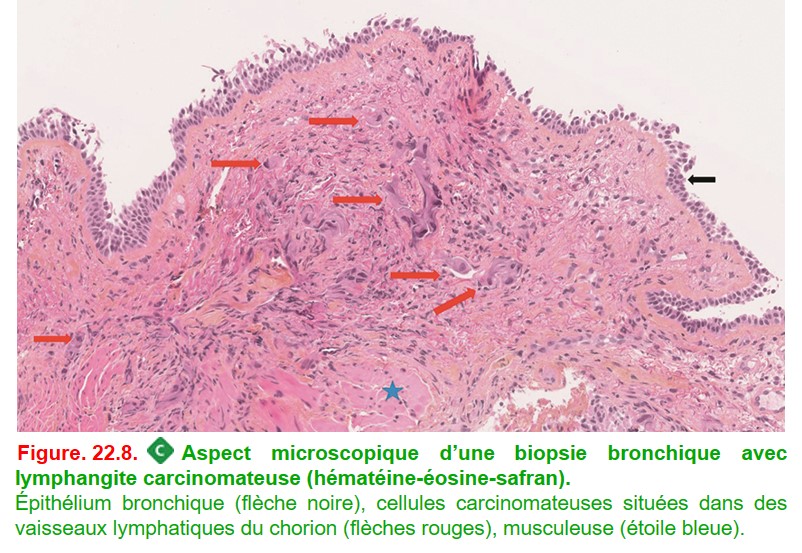
Le diagnostic de lymphangite carcinomateuse se fait sur biopsies bronchiques et transbronchiques en mettant en évidence des emboles lymphatiques tumoraux dans les septa (figure. 22.8).
{kind=link}
Les formes particulières d’adénocarcinome lépidique peuvent prendre l’aspect de PID. Dans ces cas, le LBA (± biopsie) met en évidence des cellules tumorales.
6. Lymphangio-léiomyomatose
L’aspect des lésions multikystiques diffuses, chez une femme non ménopausée, est très évocateur en radiologie. La biopsie montre en bordure de dilatations kystiques et de lymphatiques dilatés des nodules de cellules musculaires lisses immatures. Ces cellules sont considérées comme des cellules épithélioïdes périvasculaires et coexpriment l’actine du muscle lisse et un marqueur mélanocytaire, l’HMB-45 (homatropine methyle bromide-45).
7. Histiocytose langerhansienne
Le LBA montre une alvéolite macrophagique avec une proportion élevée de cellules de Langerhans mises en évidence en immunohistochimie (CD1a+). Le diagnostic de certitude est histologique mais dans un contexte clinique et scanographique compatible, une proportion de cellules CD1a+ supérieure à 5 % est fortement évocatrice du diagnostic. La biopsie (transbronchique ou chirurgicale le plus souvent) montre des amas cellulaires centrés sur les petites voies aériennes, riches en cellules de Langerhans CD1a+.
Points clés
• Les PID rassemblent de nombreuses entités dont le point commun est les opacités infiltrantes diffuses sur l’imagerie pulmonaire correspondant à une atteinte du compartiment interstitiel du poumon.
• Le diagnostic est multidisciplinaire et repose sur un faisceau d’arguments cliniques, radiologiques, biologiques et anatomopathologiques.
• Le lavage bronchoalvéolaire est un bon examen d’orientation et possiblement de certitude diagnostique. Les biopsies bronchiques et transbronchiques n’apportent des arguments diagnostiques que dans un nombre limité de PID (sarcoïdose, lymphangite). Une biopsie pulmonaire chirurgicale peut être nécessaire pour le diagnostic de pneumopathie interstitielle diffuse (le plus souvent chronique et fibrosante)
• Une analyse bactériologique et/ou minéralogique doit être prévue en fonction des hypothèses cliniques envisagées.
• Certaines lésions histologiques peuvent fortement orienter vers une étiologie (granulome épithélioïde et gigantocellulaire sans nécrose caséeuse [sarcoïdose], avec nécrose caséeuse [tuberculose], nombreuses cellules de Langerhans CD1a+ [histiocytose langerhansienne], corps asbestosiques ou silicotiques [pneumoconioses], etc.). Mais nombre de lésions ne sont pas spécifiques et nécessitent une expertise multidisciplinaire pour poser un diagnostic.
Item 206 – Épanchement pleural liquidien
Auteure : Audrey Lupo
I. Prérequis
II. Causes principales
III. Mésothéliome
Hiérarchisation des connaissances – Tableau 5
{kind=link}
I. Prérequis
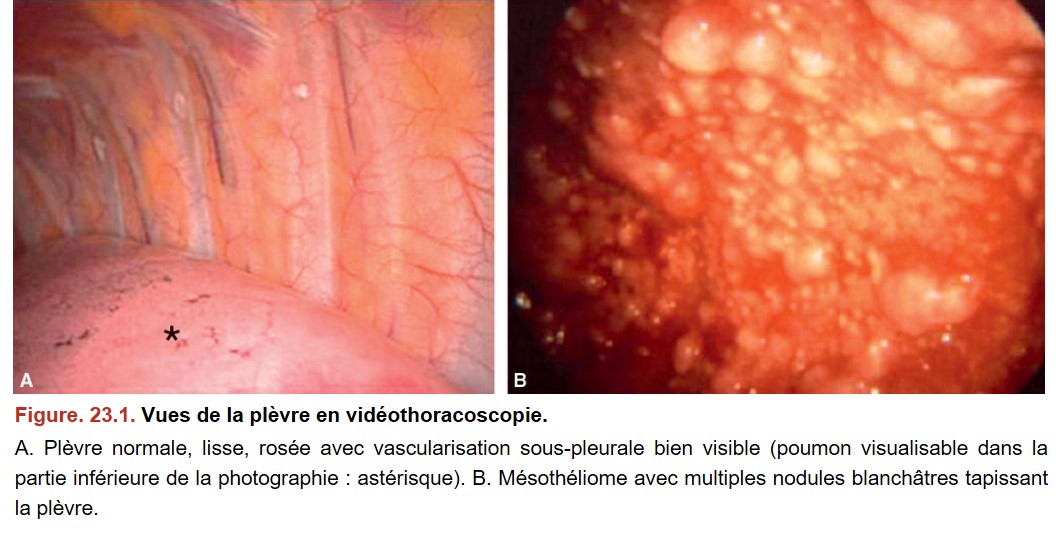
L’espace pleural est une cavité virtuelle délimitée par deux feuillets : la plèvre pariétale bordant la cavité thoracique qui se réfléchit au niveau du hile pour donner la plèvre viscérale tapissant la surface du parenchyme pulmonaire. La plèvre pariétale est divisée en trois parties en fonction des zones anatomiques qu’elle recouvre : la plèvre costale, diaphragmatique ou médiastinale. La plèvre est une séreuse comme le péritoine, le péricarde et la vaginale testiculaire. Macroscopiquement, les plèvres présentent un aspect lisse, rosé, fin, avec une vascularisation sous-pleurale bien visible (figure. 23.1A). Microscopiquement, ces plèvres sont bordées par une unique assise de cellules mésothéliales aplaties. À l’état physiologique, le liquide pleural (5 mL en permanence qui lubrifient la cavité) est principalement produit par la plèvre pariétale et réabsorbé par les lymphatiques situés entre les cellules mésothéliales de la plèvre pariétale (cela représente un débit de production/réabsorption d’environ 100 ml/j). On appelle pneumothorax la présence d’air dans la cavité pleurale et hémothorax lorsqu’il s’agit de sang. Un épanchement pleural est presque toujours pathologique, et résulte soit d’un excès de production, soit d’un défaut de réabsorption, voire des deux. Les épanchements pleuraux sont soit des transsudats (taux de protides bas < 25 g/L, de LDH bas), liés à une anomalie mécanique, soit des exsudats (taux de protides élevé [< 35 g/L], de LDH élevé [> 200 UI/L]), liés à une agression inflammatoire, infectieuse ou néoplasique.
{kind=link}
II. Causes principales
{kind=link}
III. Mésothéliome
A. Généralités
Le mésothéliome est le nom donné aux tumeurs malignes primitives des séreuses (plèvre, péritoine, péricarde et vaginale testiculaire), mais il touche le plus fréquemment la plèvre. Cette tumeur est très rare (0,3 % des cancers).
La symptomatologie est le plus souvent la survenue d’un épanchement pleural.
B. Prélèvements à visée diagnostique
1. Liquide pleural
•
• L’examen cytologique recherche des cellules malignes. Sa négativité n’élimine pas le diagnostic (sensibilité de l’ordre de 30 %).
• En cas de présence de cellules malignes, il est parfois difficile de déterminer le type des cellules tumorales (cellules adénocarcinomateuses ? cellules de mésothéliome ?).
• La cytologie seule permet d’évoquer fortement le diagnostic de mésothéliome, mais il doit être affirmé par une biopsie.
2. Biopsies pleurales pariétales avec examen anatomopathologique
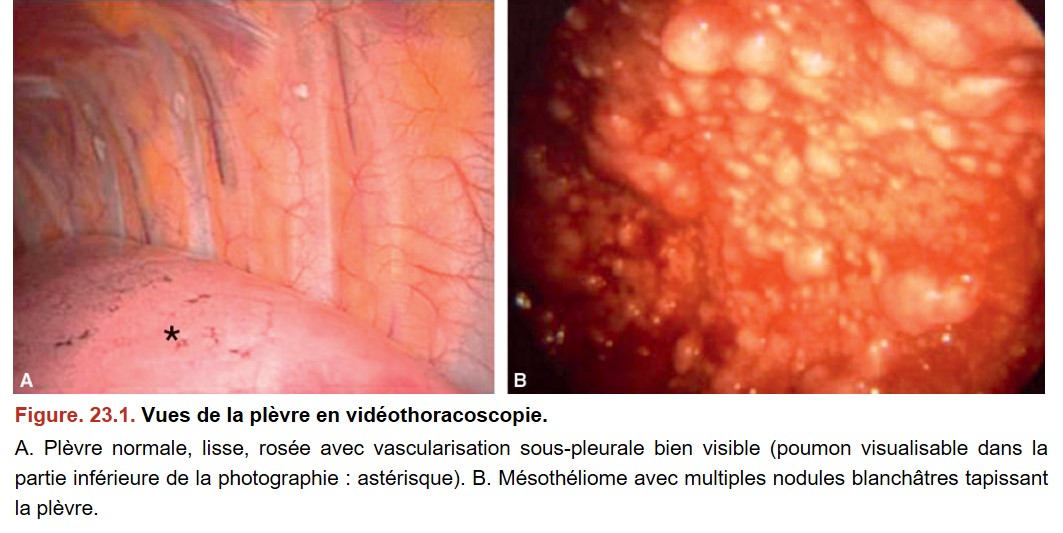{kind=link}
C. Diagnostic anatomopathologique
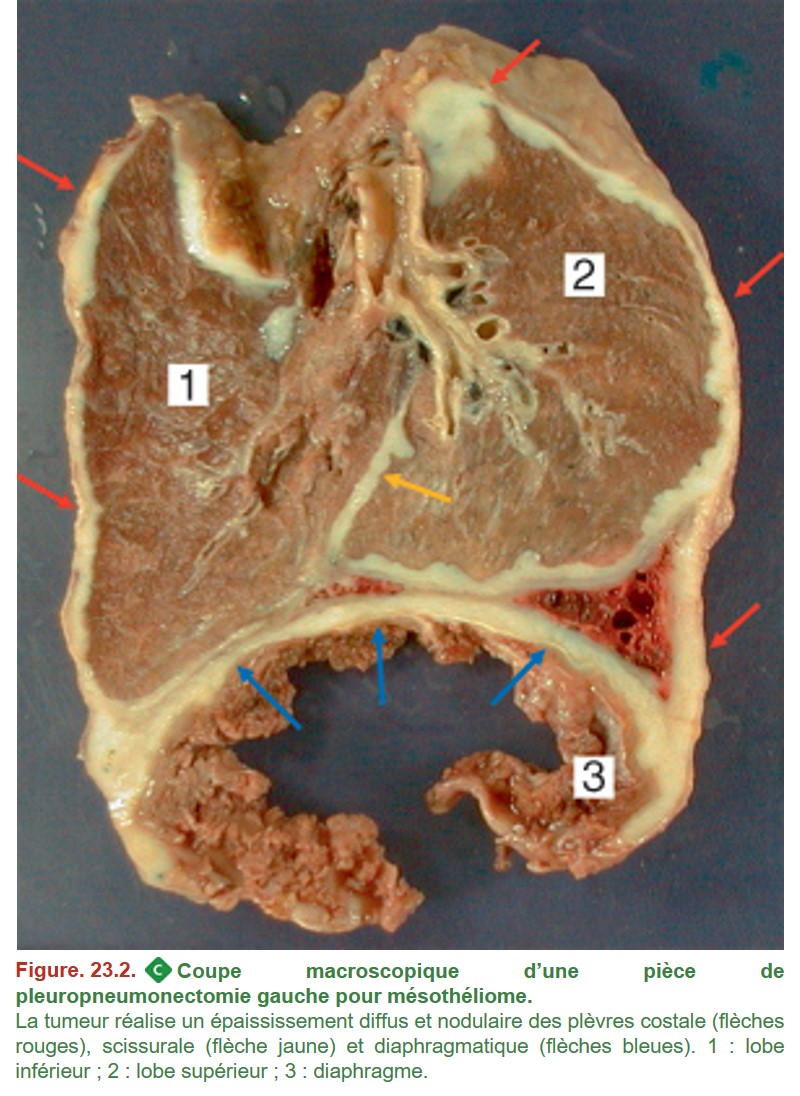
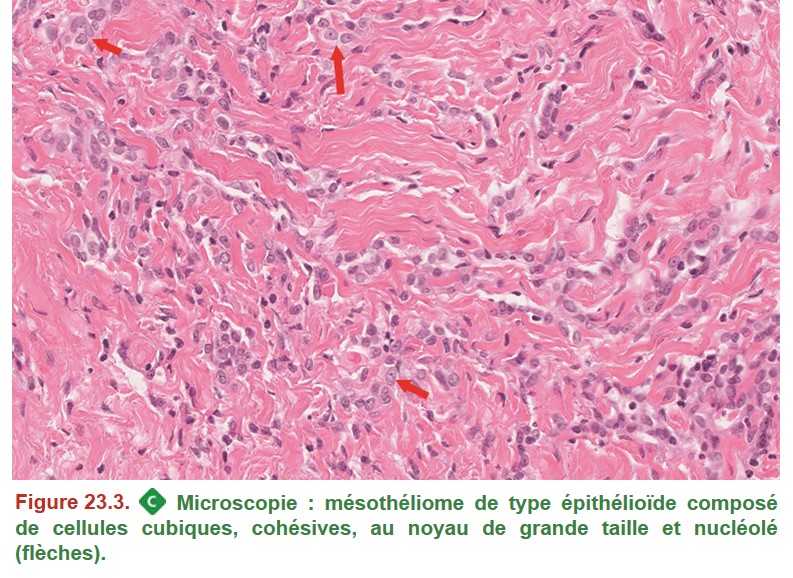
Le diagnostic de mésothéliome se porte essentiellement sur biopsie car ces tumeurs sont exceptionnellement réséquées chirurgicalement (figure. 23.2). Le diagnostic est complexe du fait de l’existence de plusieurs sous-types histologiques, notamment le sous-type épithélioïde (figure. 23.3) dont le diagnostic différentiel avec une métastase pleurale d’un carcinome est parfois difficile et nécessite obligatoirement une étude immunohistochimique pour affirmer le diagnostic.
{kind=link}
{kind=link}
De plus, la relecture par un réseau expert labellisé par l’INCa est obligatoire pour permettre une indemnisation par les fonds d’indemnisation des victimes de l’amiante (FIVA).
Points clés
Épanchement pleural
• Tout épanchement pleural doit être ponctionné, sauf si sa cause est connue, si une insuffisance cardiaque est suspectée ou si l’épanchement est minime.
• Il existe deux types d’épanchement pleural : les transsudats (plèvre saine) et les exsudats (plèvre pathologique).
Mésothéliome pleural
• Il s’agit d’une tumeur maligne rare primitive de la plèvre, survenant le plus souvent chez l’homme entre 50 et 70 ans.
• La notion d’exposition à l’amiante est fréquente mais n’est pas toujours retrouvée.
• C’est une maladie professionnelle et à déclaration obligatoire.
• Elle fait l’objet d’une demande d’indemnisation au FIVA.
• Son diagnostic nécessite une biopsie pleurale (par thoracoscopie idéalement), la cytologie du liquide pleural peut mettre en évidence des cellules malignes mais ne permet pas d’affirmer le diagnostic de mésothéliome sur ces seules données.
• Le diagnostic de mésothéliome doit être confirmé par une relecture par un groupe national d’anatomopathologistes experts.