
CHAPITRE X – Divers – Cancérologie
Plan :
• Item 297 – Cancers de l’enfant : particularités épidémiologiques, diagnostiques et thérapeutiques
• Item 299 – Tumeurs intracrâniennes
• Item 307 – Tumeurs des os primitives et secondaires
Item 297 – Cancers de l’enfant : particularités épidémiologiques, diagnostiques et thérapeutiques
Auteure : Aurore Coulomb
I. Épidémiologie et principaux cancers selon l’âge
II. Oncogenèse et syndromes de prédisposition
III. Diagnostic
IV. Principales tumeurs de l’enfant
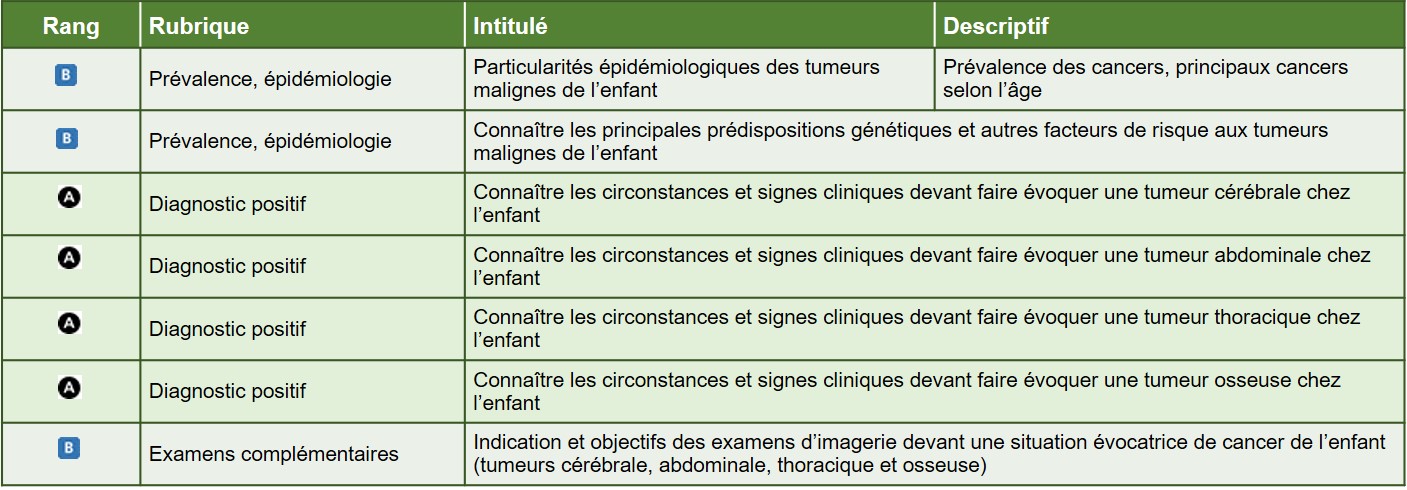
Hiérarchisation des connaissances – tableau 1
{kind=link}
I. Épidémiologie et principaux cancers selon l’âge
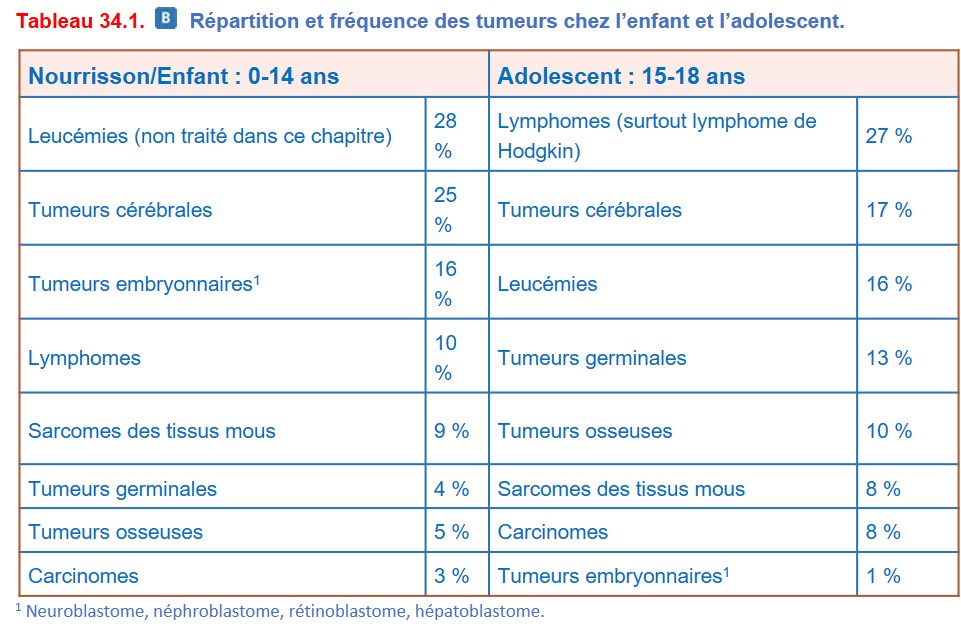{kind=link}
Le pronostic est bien meilleur que celui des cancers de l’adulte, avec des taux de guérison tous cancers confondus de l’ordre de 80 % du fait de la grande chimiosensibilité de ces cancers et de la meilleure tolérance des traitements. Cependant, des séquelles à long terme (maladies chroniques, second cancer) nécessitent une surveillance des patients sur une longue durée. Les cancers pédiatriques représentent la deuxième cause de mortalité chez l’enfant de plus de 1 an en France, après les causes accidentelles.
II. Oncogenèse et syndromes de prédisposition
Les cancers pédiatriques se développent souvent à partir de cellules embryonnaires persistant après la naissance ou bien de cellules indifférenciées. Un changement de la séquence de l’ADN de gènes de ces cellules peut les transformer en cellules cancéreuses qui vont se diviser et diffuser dans l’organisme. L’identification de certaines mutations permet dans certains cas d’administrer des traitements ciblés. Une faible proportion de ces cancers (< 10 %) rentre dans le cadre de syndromes de prédisposition génétique : mutation germinale du gène Rb (rétinoblastome), syndrome de Li-Fraumeni (mutation germinale de p53), syndromes chromosomiques (trisomie 21, syndrome de Klinefelter, etc.), anomalies du développement : syndrome de Wiedemann-Beckwith (néphroblastome et hépatoblastome), neurofibromatoses de type 1 ou 2, néoplasie endocrinienne multiple, etc. Les facteurs environnementaux sont très rarement impliqués chez l’enfant (irradiations ou traitement par Distilbène (diethylstilbestrol) chez la femme enceinte, virus Epstein-Barr pour le lymphome de Burkitt, virus de l’hépatite B pour le carcinome hépatocellulaire ou VIH dans certains lymphomes).
III. Diagnostic
L’examen ACP permet :
• de faire le diagnostic ;
• d’évaluer le pronostic à partir de critères propres à chaque tumeur ;
• d’évaluer la régression tumorale en cas de chimiothérapie préopératoire (quantification des remaniements post-chimiothérapiques témoignant d’une efficacité thérapeutique).
En France, toute tumeur survenant chez un sujet de moins de 18 ans doit systématiquement faire l’objet de prélèvements à l’état frais pour être congelés (cryopréservation obligatoire avec conservation dans les tumorothèques/centres de ressources biologiques). Il en est de même pour les cellules en cas de leucémie (recommandations INCa, 2011). Pour les leucémies, le diagnostic est établi par l’analyse du myélogramme. En raison de la rareté de ces tumeurs et de la spécificité de leur prise en charge, leur diagnostic définitif doit être fait de manière concertée dans un centre spécialisé en oncologie pédiatrique.
IV. Principales tumeurs de l’enfant
A. Tumeurs hématopoïétiques
Leurs caractéristiques sont détaillées dans le tableau 34.2.
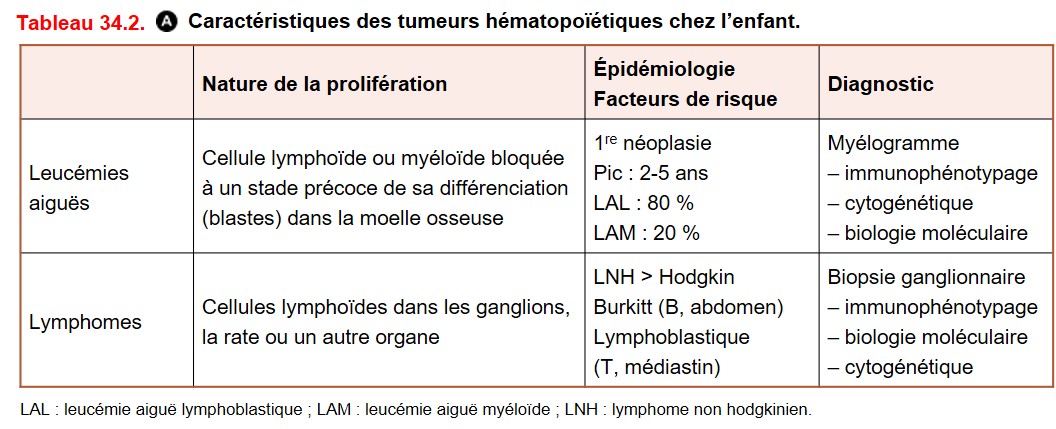{kind=link}
1. Leucémie
C’est une prolifération clonale développée à partir d’une cellule lymphoïde (LAL pour leucémie aiguë lymphoblastique) ou myéloïde (LAM pour leucémie aiguë myéloïde) bloquée à un stade précoce de sa différenciation dans la moelle osseuse. Le diagnostic est avant tout hématologique par la mise en évidence de blastes sur le myélogramme (> 20 %).
2. Lymphome
C’est une prolifération tumorale de cellules lymphoïdes dans les ganglions lymphatiques, la rate ou un autre organe (lymphome extraganglionnaire). On distingue le lymphome de Hodgkin (LH) des lymphomes non hodgkiniens (LNH).
• le lymphome de Burkitt de phénotype B, le plus souvent de siège abdominal (environ 50 % des cas) ;
• le lymphome lymphoblastique, le plus souvent de siège médiastinal, de phénotype T (environ 30 % des cas).
B. Tumeurs du système nerveux cérébral
Les tumeurs cérébrales sont les tumeurs solides les plus fréquentes de l’enfant et la 2e cause de cancer après les tumeurs hématopoïétiques. Le pic d’incidence se situe entre 3 et 5 ans. Les tumeurs cérébrales de l’enfants sont principalement sous-tentorielles (fosse postérieure). Parmi ces tumeurs, les plus fréquentes sont les tumeurs gliales (astrocytome pilocytique), le médulloblastome, l’épendymome. Le diagnostic de ces tumeurs repose sur la réalisation d’une biopsie stéréotaxique avec examen anatomopathologique. Le pronostic vital et fonctionnel, très variable, dépend avant tout du type histologique, de la localisation (et donc de sa potentielle exérèse chirurgicale complète) et de l’âge.
•
• Le médulloblastome est une tumeur cérébrale hautement maligne se développant dans le cervelet. Les cellules tumorales sont très peu différenciées et ressemblent aux neuroblastes du tube neural de l’embryon.
• L’épendymome est une tumeur le plus souvent bénigne dérivée des cellules épendymaires (qui tapissent l’intérieur des cavités contenant du liquide cérébrospinal).
C. Tumeurs du blastème d’organe
{kind=link}
Le neuroblastome est une prolifération tumorale des cellules embryonnaires du système nerveux autonome sympathique, dérivées de la crête neurale. La plupart sont développés dans la médullosurrénale (même origine embryologique), les autres le long de la chaîne des ganglions orthosympathiques. Le diagnostic est avant tout biologique par la mise en évidence d’une augmentation des catécholamines urinaires, et d’une fixation sur la scintigraphie à la MIBG (méta-iodobenzylguanidine).
D. Tumeurs malignes conjonctives : sarcomes
• le rhabdomyosarcome : c’est une tumeur à différenciation musculaire striée avec deux pics d’incidence : le premier entre 1 et 5 ans et le second entre 15 et 19 ans,
• l’ostéosarcome : c’est la tumeur osseuse maligne la plus fréquente chez l’enfant et l’adolescent avec un âge moyen de 14 ans (pic à la puberté), et une atteinte préférentielle près du genou.
Sarcome d’Ewing : C’est la 2e cause de tumeurs osseuses malignes après les ostéosarcomes. Il s’agit d’une tumeur maligne très peu différenciée à fort potentiel métastatique caractérisée par une translocation spécifique t(11;22)(q24 ; q12) responsable de la formation d’un transcrit de fusion le plus souvent EWSR-FLI-1, détecté par FISH et par séquençage de l’ARN (tableau 34.4). D’autres fusions impliquant d’autres gènes de la famille ETS ou non-ETS ont également été décrites.
{kind=link}
E. Tumeurs germinales malignes gonadiques ou extragonadiques
{kind=link}
Points clés
• Les tumeurs sont rares chez l’enfant (1 à 2 % des cancers) mais représentent la 2e cause de mortalité après l’âge de 1 an en France, après les accidents.
• Les types histologiques sont très distincts de ceux de l’adulte (les carcinomes sont rares, les tumeurs hématopoïétiques et tumeurs cérébrales plus fréquentes).
• La fréquence des tumeurs est différente chez l’enfant et l’adolescent.
– chez l’enfant, les plus fréquentes sont les leucémies et les tumeurs cérébrales ;
– chez l’adolescent, les plus fréquentes sont les lymphomes et les tumeurs germinales.
• La prise en charge multidisciplinaire en centre spécialisé est indispensable.
• Des analyses complémentaires de biologie moléculaire et génomiques pour identifier un transcrit de fusion ou une amplification de gène complètent les investigations habituelles.
• Toute tumeur survenant chez un sujet de moins de 18 ans doit faire l’objet de prélèvements à l’état frais (sans fixateur) pour être congelés car les analyses moléculaires sont parfois possibles uniquement à partir de tissu congelé.
Item 299 – Tumeurs intracrâniennes
Auteur : Franck Bielle
I. Prérequis : rappels histologiques
II. Types histologiques des tumeurs primitives du système nerveux central
III. Métastases cérébrales
IV. Circonstances de découverte
V. Spécificités des tumeurs intracrâniennes chez l’enfant
Hiérarchisation des connaissances – Tableau 2
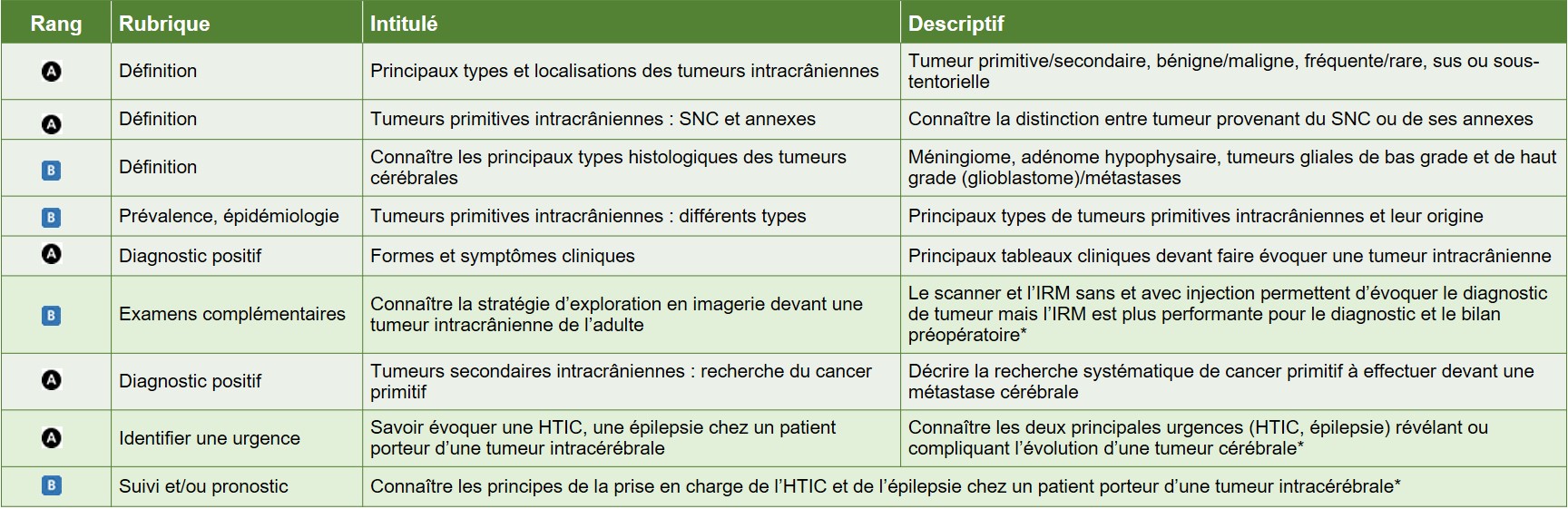{kind=link}
I. Prérequis : rappels histologiques
Le parenchyme du système nerveux central (SNC) est constitué de neurones et de cellules gliales. Les cellules gliales sont réparties en deux familles :
• les cellules macrogliales provenant du neurectoderme :
– les astrocytes : cellules étoilées régulant le métabolisme des neurones,
– les oligodendrocytes : élaboration des gaines de myéline,
– les cellules épendymaires bordant les ventricules. Les plexus choroïdes sont constitués de cellules épendymaires spécialisées et de vaisseaux, et produisent le liquide cérébrospinal ;
• les cellules microgliales, qui sont les macrophages résidents du SNC.
Les méninges sont constituées de la dure-mère (au contact de l’os), de l’arachnoïde, puis de la pie-mère adhérant au tissu nerveux. Le liquide cérébrospinal (LCS) circule dans les ventricules, puis dans l’espace sous-arachnoïdien.
II. Types histologiques des tumeurs primitives du système nerveux central
{kind=link}
Les tumeurs primitives sont issues de tissus intracrâniens tandis que les métastases sont issues de la dissémination intracrânienne d’une tumeur prenant son origine dans un autre organe. Le grade des tumeurs primitives, suivant la classification de l’OMS, s’étend de 1 (bénin) à 4 (haute malignité). Les tumeurs primitives de haut grade (3 et 4) métastasent rarement dans les autres organes (foie, poumons, os), mais peuvent disséminer dans le SNC via le LCS. On distingue les tumeurs :
• extra-parenchymateuses (développée à partir des annexes du SNC comme les méninges ou les nerfs crâniens) ;
• intraparenchymateuses (dans le parenchyme du SNC) ;
• intraventriculaires.
On distingue les topographies :
• supratentorielle (au-dessus de la tente du cervelet), 70 % des tumeurs de l’adulte ;
• infratentorielle (en dessous de la tente du cervelet), 30 % des tumeurs de l’adulte.
Les tumeurs intracrâniennes les plus fréquentes sont les métastases. Les tumeurs intracrâniennes primitives les plus fréquentes sont développées à partir des annexes du SNC : ce sont les méningiomes et les adénomes hypophysaires. Les tumeurs intracrâniennes primitives les plus fréquentes développées à partir du parenchyme du SNC sont les glioblastomes.
L’ordre décroissant de fréquence est donc : métastases > méningiomes et adénomes hypophysaires > glioblastomes.
A. Gliomes diffus
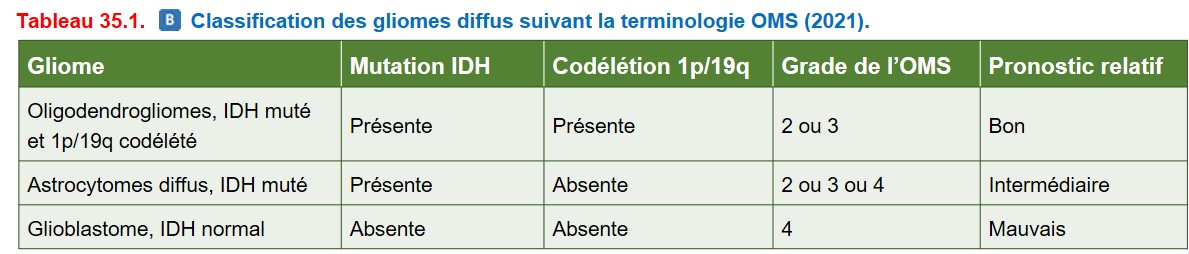
Ces tumeurs gliales infiltrent le tissu nerveux sous la forme de cellules tumorales isolées à distance du centre de la tumeur, ce qui empêche leur résection complète. Elles sont d’évolution létale. Les tumeurs de grade 2 (bas grade de malignité) progressent inexorablement vers des lésions de grade 3 ou 4 (hauts grades de malignité). Les cellules tumorales ressemblent à des astrocytes et/ou des oligodendrocytes. Le diagnostic intègre des critères histologiques et moléculaires et permet d’identifier des pronostics très différents (tableau 35.1) :
{kind=link}
• les oligodendrogliomes (avec mutation IDH et délétion des bras chromosomiques 1p et 19q) sont de grade 2 ou 3 et présentent les survies les plus longues ;
• les astrocytomes diffus (avec mutation IDH) présentent un pronostic intermédiaire, sont de grade 2, 3 ou 4 (le grade est augmenté au cours de l’évolution) ;
• les glioblastomes sans mutation IDH sont de grade 4, ce sont les gliomes diffus les plus fréquents de l’adulte et présentent un mauvais pronostic.
On recherche également une hyperméthylation du promoteur du gène de la MGMT, qui prédit la sensibilité à la chimiothérapie.
B. Gliomes circonscrits
Par opposition aux gliomes diffus, les gliomes circonscrits présentent une bonne délimitation par rapport au tissu nerveux. Ils sont le plus souvent bénins (de grade 1). Le gliome circonscrit le plus fréquent est l’astrocytome pilocytique qui survient chez l’enfant, généralement au niveau du cervelet ou du tronc cérébral. Il a une croissance lente et le traitement repose sur une exérèse chirurgicale complète qui permet la guérison.
C. Tumeurs neuronales et glioneuronales
D. Tumeurs épendymaires et dérivées des plexus choroïdes
• Tumeurs épendymaires (apparentées aux épendymocytes qui bordent les ventricules) :
– subépendymome (grade 1, bénin), souvent dans le 4e ventricule ;
– épendymomes (grade 2 ou 3) supratentoriels, ou de la fosse postérieure ou de la moelle spinale.
• Tumeurs des plexus choroïdes :
– papillome des plexus choroïdes (grade 1) ;
– papillome atypique des plexus choroïdes (grade 2) ;
– carcinome des plexus choroïdes (grade 3).
E. Médulloblastome
C’est la tumeur embryonnaire la plus fréquente du SNC. Elle survient surtout chez l’enfant et parfois chez l’adulte jeune. Elle est maligne, de grade 4, et prend son origine par définition dans le cervelet. Le risque de dissémination métastatique dans le LCS impose au diagnostic initial une IRM pan médullaire et une ponction lombaire en l’absence de contre-indication.
F. Méningiomes
Ce sont des tumeurs extra-parenchymateuses fréquentes, le plus souvent bénignes et développées à partir des cellules arachnoïdiennes dans les méninges. Les méningiomes représentent un tiers des tumeurs primitives cérébrales opérées (sex-ratio = 2 femmes pour 1 homme, avec un pic de fréquence se situant entre 50 et 70 ans). Les formes symptomatiques ou montrant une évolutivité radiologique sont une indication à un traitement chirurgical et/ou par radiothérapie. La découverte fortuite d’un méningiome de petite taille sans indication opératoire est aussi habituelle. Les facteurs prédisposants de méningiomes avec possibilité de localisations multiples sont :
• génétiques : neurofibromatose de type 2 ;
• hormonaux : traitement progestatif ;
• les radiations ionisantes. Il s’agit de la tumeur cérébrale radio-induite la plus fréquente.
L’OMS distingue trois grades pronostiques de méningiomes :
• méningiome (grade 1, bénin) ;
• méningiome atypique (grade 2) ;
• méningiome malin (grade 3).
G. Adénomes hypophysaires
Les adénomes hypophysaires sont très fréquents et sont observés de façon fortuite dans 20 % de la population. Les formes symptomatiques peuvent être sécrétantes (présence de symptômes en rapport avec la production excessive d’hormones hypophysaires : prolactine, GH, LH, FSH, ACTH [growth, luteinizing, folliculo-stimulating et adrenocorticotrophic hormones respectivement] ou TSH) ou non sécrétantes. La taille est très variable entre les micro-adénomes (< 10 mm) et les macro-adénomes (> 10 mm). Les adénomes hypophysaires peuvent causer :
• un syndrome endocrinien : hypersécrétion d’un secteur hormonal, et/ou insuffisance d’un ou plusieurs secteurs hormonaux anté ou post-hypophysaires, voire panhypopituitarisme ;
• un syndrome de masse : compression du chiasma optique (hémianopsie bitemporale), céphalées ;
• une apoplexie hypophysaire (nécrose hémorragique d’expansion rapide responsable d’un tableau clinique aigu et grave avec hypertension intracrânienne).
H. Schwannomes
Ces tumeurs se développent à partir des cellules de Schwann des nerfs crâniens et périphériques (à l’exception des nerfs olfactifs et du nerf optique). Elles sont bénignes (grade 1) et surviennent de façon sporadique ou dans le cadre de la neurofibromatose de type 2. La localisation préférentielle est le nerf vestibulaire (méat acoustique interne et angle pontocérébelleux).
I. Lymphomes primitifs du système nerveux central
Ils correspondent principalement aux lymphomes B diffus à grandes cellules, représentent 2 à 3 % des tumeurs cérébrales, et surviennent chez le sujet âgé sous forme de lésions multiples périventriculaires. Chez le sujet jeune, ce diagnostic doit faire rechercher une immunodépression.
J. Tumeurs germinales intracérébrales
Elles se divisent selon les mêmes sous-types histologiques que les localisations gonadiques.
K. Craniopharyngiome
Il s’agit d’une tumeur épithéliale, souvent kystique et calcifiée, bénigne mais d’exérèse complète parfois difficile et avec un risque de récidive locale. Elle se développe à partir des restes embryonnaires de la poche de Rathke, dans et au-dessus de l’hypophyse, chez l’enfant et l’adulte.
III. Métastases cérébrales
Le diagnostic histologique d’une métastase repose sur :
• la nature (lignée cellulaire) de la composante tumorale maligne (tumeur épithéliale maligne = carcinome ; tumeur mélanocytaire maligne = mélanome, etc.) ;
• l’expression de certaines protéines et/ou la sécrétion de certaines substances par les cellules tumorales, mises en évidence le plus souvent par immunohistochimie ou colorations spéciales (récepteurs hormonaux, mucines, marqueurs neuroendocrines). Le choix des immunohistochimies est fonction des hypothèses diagnostiques les plus probables et également de l’incidence thérapeutique éventuelle.
Le prélèvement neurochirurgical (biopsie ou exérèse) d’une métastase permet donc :
• le diagnostic histologique de la tumeur (ex : adénocarcinome) ;
• l’orientation vers un primitif (origine mammaire ?) ;
• l’identification éventuelle de cibles thérapeutiques (récepteurs hormonaux, HER2, PD-L1, recherche de mutations prédictives de réponse ou non-réponse à des traitements dits « ciblés ») ;
• en cas d’exérèse, le traitement rapide de l’effet de masse.
IV. Circonstances de découverte
• de céphalées pouvant s’intégrer dans un syndrome d’hypertension intracrânienne ;
• de crises comitiales ;
• de déficits neurologiques focaux plus ou moins rapidement progressifs ;
• de troubles psychiatriques (troubles du comportement, troubles de l’humeur).
Dans tous les cas, le diagnostic de tumeur est suspecté sur les données de l’imagerie sans et avec injection de produit de contraste (IRM plus performante que le scanner) mais ne peut être affirmé que par l’examen histologique de biopsies cérébrales ou d’une pièce d’exérèse de la lésion.
{kind=link}
• Les diagnostics différentiels peuvent être :
• des lésions infectieuses (abcès cérébral, toxoplasmose cérébrale [sujet VIH], tuberculome, etc.) ;
• éventuellement une malformation artérioveineuse, un accident vasculaire ischémique ou hémorragique, une maladie inflammatoire (sclérose en plaques, sarcoïdose, etc.).
V. Spécificités des tumeurs intracrâniennes chez l’enfant
Points clés
• Les métastases cérébrales sont les tumeurs intracrâniennes les plus fréquentes chez l’adulte devant les tumeurs cérébrales primitives.
• En cas de métastases, le primitif est le plus souvent un cancer bronchopulmonaire (30 %), un carcinome mammaire (25 %), rénal (7 %), digestif (7 %) ou un mélanome (7 %).
• Si un cancer primitif évolutif (avec preuve histologique) est connu, les métastases cérébrales ne sont en général pas biopsiées.
• Chez l’adulte, plus de la moitié des tumeurs primitives intracrâniennes sont bénignes (méningiomes bénins, adénomes hypophysaires). Cependant, la tumeur primitive intraparenchymateuse la plus fréquente est le glioblastome sans mutation IDH, grade 4, de mauvais pronostic.
• Les tumeurs malignes intracrâniennes primitives ne donnent qu’exceptionnellement des métastases systémiques (retenir qu’elles restent dans le SNC ou ses enveloppes).
• Chez l’adulte, 70 % des tumeurs sont de topographie supratentorielle ; chez l’enfant elles sont dans 70 % des cas sous-tentorielles.
• Le diagnostic d’une tumeur primitive cérébrale repose sur l’examen histologique.
Item 307 – Tumeurs des os primitives et secondaires
Auteure : Frédérique Larousserie
I. Généralités
II. Tumeurs osseuses secondaires
II. Tumeurs osseuses primitives
Hiérarchisation des connaissances – Tableau 3
{kind=link}
I. Généralités
A. Diagnostic – Prélèvements
• donne des arguments en faveur de l’agressivité d’une lésion osseuse (radiographies, TDM) ;
• permet une orientation diagnostique en fonction de l’aspect, de la localisation, du terrain. Dans quelques cas, elle identifie certaines lésions bénignes qui ne nécessitent pas un geste biopsique ou chirurgical ;
• est nécessaire au bilan d’extension locale en cas d’indication chirurgicale (IRM). L’IRM permet de déterminer avec précision les limites de la tumeur et ses rapports avec les structures adjacentes.
La biopsie osseuse avec examen anatomopathologique est nécessaire pour établir le diagnostic, sauf pour les métastases osseuses d’un cancer primitif connu évolutif ou pour les tumeurs bénignes d’aspect radiologique typique (chondromes des extrémités [phalanges, métacarpiens], fibromes non ossifiants, exostoses typiques des membres, dysplasies fibreuses typiques, kystes osseux essentiels, ostéome ostéoïde typique).La biopsie peut être chirurgicale ou réalisée au trocart avec guidage par imagerie (TDM). Les biopsies des tumeurs de l’enfant et/ou avec suspicion de sarcome doivent également faire l’objet d’une cryopréservation d’un fragment tumoral frais (non fixé) pour d’éventuelles études moléculaires ultérieures à visée diagnostique ou pronostique (recommandations INCa, 2011). Les prélèvements osseux devront le plus souvent être décalcifiés (décalcification à l’EDTA [éthylène-diamine-tétra-acétique] ou aux acides faibles), ce qui allonge le délai de réponse.
Les principaux diagnostics différentiels d’une tumeur osseuse sont :
• un cal osseux (processus cicatriciel), notamment sur fracture de fatigue (sans traumatisme) ;
• une infection (ostéomyélite, abcès, ostéite chronique) ;
• un infarctus osseux.
B. Différents types de tumeurs osseuses
1. Histologie des tumeurs osseuses primitives : généralités
La classification des tumeurs primitives des os selon l’OMS, publiée en 2020, dénombre environ 50 entités tumorales bénignes et malignes.
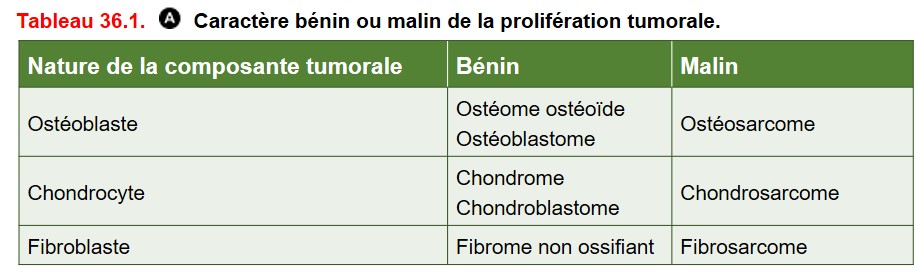
La dénomination de ces tumeurs repose sur :
• la lignée de différenciation de la cellule tumorale. Exemples :
– ostéoblaste : ostéoblastome, ostéosarcome,
– chondrocyte : chondrome, chondrosarcome ;
• le caractère bénin ou malin de la prolifération tumorale (tableau 36.1) :
{kind=link}
– ostéoblastome : tumeur bénigne,
– ostéosarcome : tumeur maligne,
– sarcome : tumeur mésenchymateuse maligne.
Remarque : Certaines tumeurs bénignes peuvent se transformer en tumeur maligne, principalement les tumeurs cartilagineuses.
Les biopsies sont préférentiellement chirurgicales en cas de suspicion de tumeur osseuse primitive non hématopoïétique, car ces tumeurs sont souvent hétérogènes. La biopsie doit être faite dans un centre spécialisé (dans le réseau labellisé par l’INCa ResOs) car ce sont des tumeurs rares dont la prise en charge diagnostique et thérapeutique nécessite une équipe multidisciplinaire réunissant radiologues, chirurgiens et anatomopathologistes expérimentés. La biopsie doit être faite de préférence par le chirurgien qui prendra ensuite en charge le patient pour le traitement chirurgical ultérieur éventuel : le trajet de la biopsie, qui doit être enlevé avec la tumeur osseuse primitive en cas de malignité en raison du risque d’essaimage tumoral le long du trajet, doit respecter certaines règles techniques afin de minimiser la contamination des tissus sains et de ne pas compromettre la possibilité d’une chirurgie conservatrice dans un deuxième temps. Si une biopsie au trocart est choisie, la voie d’abord de la biopsie faite par le radiologue est discutée avec le chirurgien qui prendra en charge le patient pour le traitement ultérieur. Les images histologiques s’interprètent en fonction de la clinique (âge, antécédents, lésion osseuse préexistante) et de l’imagerie +++.
2. Histologie des tumeurs secondaires (métastases) : généralités
• le diagnostic histologique de la tumeur (ex : adénocarcinome) ;
• assez souvent, mais pas dans tous les cas, l’orientation vers un primitif (ex : origine mammaire) ;
• l’identification éventuelle de cibles thérapeutiques, par exemple :
– dans le cas d’un adénocarcinome d’origine mammaire, recherche de l’expression des récepteurs hormonaux et de HER2 par les cellules tumorales par étude immunohistochimique,
– dans la métastase d’un primitif pulmonaire, recherche de mutations prédictives de réponse ou non-réponse à des traitements dits « ciblés ».
Le diagnostic histologique d’une métastase repose sur :
• la lignée cellulaire d’origine de la composante tumorale maligne (tumeur épithéliale maligne = carcinome ; tumeur mélanocytaire maligne = mélanome, etc.) ;
• l’expression de certaines protéines et/ou la sécrétion de certaines substances par les cellules tumorales, mises en évidence le plus souvent par immunohistochimie ou histochimie (récepteurs hormonaux, mucines, granules sécrétoires neuroendocrines, etc.).
Le choix des anticorps pour une étude immunohistochimique complémentaire dépend des hypothèses diagnostiques faites sur l’aspect morphologique de la tumeur observée sur la coloration standard (HE ou HES), sur les renseignements cliniques (antécédents carcinologiques), et également de l’incidence thérapeutique éventuelle.
II. Tumeurs osseuses secondaires
•
• Os = 3e site métastatique le plus fréquent après le foie et le poumon.
• Type histologique le plus fréquent : carcinomes.
• Cancers les plus ostéophiles : sein, prostate, thyroïde, rein, poumon.
Les métastases osseuses peuvent être inaugurales (révélatrices) ou non. Les métastases sont d’aspect radiologique très varié.
• le diagnostic histologique de la lésion (métastase d’un carcinome ou d’un mélanome ou leurs diagnostics différentiels : myélome, lymphome, sarcome, autres) ;
• l’orientation vers un primitif lorsqu’il n’est pas connu ou retrouvé ;
• l’identification éventuelle de cibles thérapeutiques (récepteurs hormonaux, HER2, recherche de mutations prédictives de réponse ou non-réponse à des traitements dits « ciblés »).
La preuve histologique (certitude diagnostique) est nécessaire car :
• les traitements sont potentiellement toxiques ;
• ils dépendent des types histologiques et du cancer primitif ;
• certains sont « ciblés » (c’est-à-dire qu’ils ne sont efficaces qu’en présence de l’expression de certaines protéines ou qu’en présence ou non de certaines mutations). Il peut arriver que l’on ne retrouve pas le primitif (environ 10 % des cas). Il s’agit en général d’adénocarcinomes dits alors de « primitif inconnu » (adenocarcinoma of unknown primary [ACUP]) et ils sont de mauvais pronostic.
III. Tumeurs osseuses primitives
A. Tumeurs osseuses bénignes
Les tumeurs osseuses bénignes peuvent se comporter de façon variable : certaines sont quiescentes, d’autres actives, et enfin certaines sont agressives localement.
1. Ostéome ostéoïde
• Histologie : il s’agit d’une tumeur ostéoformatrice bénigne avec néotravées d’os immature courtes et anastomosées, bordées par des ostéoblastes et des ostéoclastes, et avec des espaces entre les travées richement vascularisés.
• Relativement fréquent, patient jeune (enfant, adolescent, jeune adulte).
• Imagerie typique en radiographie standard et TDM avec « nidus » : lacune claire avec condensation centrale, de taille < 2 cm, entourée par une sclérose osseuse réactionnelle (TDM) et par de l’œdème (IRM).
• Le plus souvent, biopsie non nécessaire au diagnostic.
• Traitement de choix : thermoablation par radiofréquence ou cryothérapie, avec examen histologique sur biopsie au trocart faite dans le même temps pour confirmation diagnostique (confirmation possible dans 30 à 60 % des cas seulement).
2. Ostéochondrome (également dénommée exostose ostéogénique)
• Tumeur ostéocartilagineuse développée à la surface de l’os, métaphyse des os longs, os plats, pédiculée ou sessile.
• Histologie : coiffe cartilagineuse hyaline peu cellulaire de moins de 2 cm d’épaisseur maximale, non nodulaire, donnant naissance, par ossification enchondrale, à un tissu osseux d’architecture normale.
• Diagnostic fait sur l’imagerie +++ (radiographie standard ou TDM : continuité corticomédullaire +++, IRM : coiffe cartilagineuse en hypersignal, d’épaisseur maximale de 2 cm, non nodulaire).
• Biopsie inutile, abstention ou traitement chirurgical en présence de symptômes ou en cas de suspicion de transformation en chondrosarcome (dans ce cas, pas de biopsie mais résection en bloc d’emblée).
• Formes multiples : maladie des exostoses multiples.
• Possibilité de dégénérescence en chondrosarcome (dénommé chondrosarcome périphérique) : de 1 % (ostéochondrome solitaire) à 5 % (maladie des exostoses multiples).
3. Tumeur cartilagineuse bénigne : chondrome
• Histologie : tumeur constituée de nodules de cartilage hyalin, peu cellulaires, bien limités et séparés par de la moelle adipeuse ou hématopoïétique.
• Le plus souvent, diagnostic fait sur l’imagerie, biopsie non nécessaire, abstention ou traitement chirurgical en présence de symptômes ou en cas de suspicion de transformation en chondrosarcome de bas grade (curetage d’un enchondrome).
• Formes multiples : enchondromes multiples dans les enchondromatoses (maladie d’Ollier et syndrome de Maffucci).
• Possibilité de dégénérescence en chondrosarcome fréquente (entre 40 et 50 %) en cas d’enchondromes multiples.
4. Fibrome non ossifiant (corticomédullaire)/cortical defect (quand la lésion est limitée à la corticale)
• Histologie : cellules fibroblastiques organisées en faisceaux courts entrecroisés, mêlées à des lymphocytes et à des macrophages sur un fond fibreux.
• Enfant, tumeur très fréquente.
• Formes typiques diagnostiquées par le radiologue sur radiographie standard.
5. Dysplasie fibreuse
• Histologie : lésion fibro-osseuse avec un contingent osseux sous la forme de néotravées d’os immature de forme variée (classiquement décrite comme alphabétique), sur un fond fibreux avec petites cellules non atypiques (préostéoblastes). Mutation du gène GNAS retrouvée dans environ 70 % des cas.
• Lésion fréquente.
• À tout âge, découverte chez l’enfant et l’adulte jeune.
• Lésion unique (70 %) ou multiple (30 %).
• Os longs, os du crâne et de la face, côtes.
• Signes cliniques : déformation osseuse, fracture pour les os longs, douleur.
• Diagnostic par imagerie (radiographie standard ± TDM) : lésion bien limitée, avec matrice en verre dépoli.
6. Tumeur à cellules géantes de l’os
• Histologie : tumeur riche en cellules géantes de type ostéoclastique (cellules non tumorales de la lignée monocyte-macrophage, responsables de la lyse osseuse) harmonieusement réparties sur un fond de petites cellules stromales préostéoblastiques sans atypies (cellules tumorales). Mutation du gène H3F3A avec expression nucléaire de la protéine mutée H3.3 G34W (la plus fréquente) dans environ 90 % des cas en immunohistochimie, le plus souvent conservée en cas de transformation sarcomateuse. Les cellules stromales expriment la molécule RANK ligand (RANKL : receptor activator of nuclear factor kappa-B ligand), qui permet le recrutement, la prolifération et l’activation des cellules géantes de type ostéoclastique, celles-ci exprimant le RANK.
• Adulte jeune (pas dans le squelette immature).
• Tumeur dite intermédiaire car agressive localement avec récidives fréquentes.
• Parfois implants pulmonaires considérés comme « bénins ».
• Localisation métaphysoépiphysaire +++ sur les os longs, atteinte possible des os plats et courts.
• Tumeur lytique excentrée avec déformation des contours de l’os, limitée par une fine coque osseuse (pas de réelle infiltration des tissus mous).
• Dégénérescence en sarcome (ostéosarcome ou sarcome indifférencié) rare (tumeur à cellules géantes maligne).
• Traitement chirurgical (curetage-comblement, parfois itératif ; dans certains cas résection).
• Dénosumab dans les formes inopérables : anticorps anti-RANKL qui empêche la formation du complexe RANK-RANKL à la surface des cellules géantes de type ostéoclastique et permet d’inhiber la lyse osseuse, de favoriser la minéralisation de la tumeur sans toutefois éliminer complètement les cellules tumorales.
7. Autres tumeurs primitives bénignes
Kyste osseux essentiel (enfants), kyste osseux anévrismatique (enfants, adolescents), angiome, etc.
B. Tumeurs osseuses primitives malignes
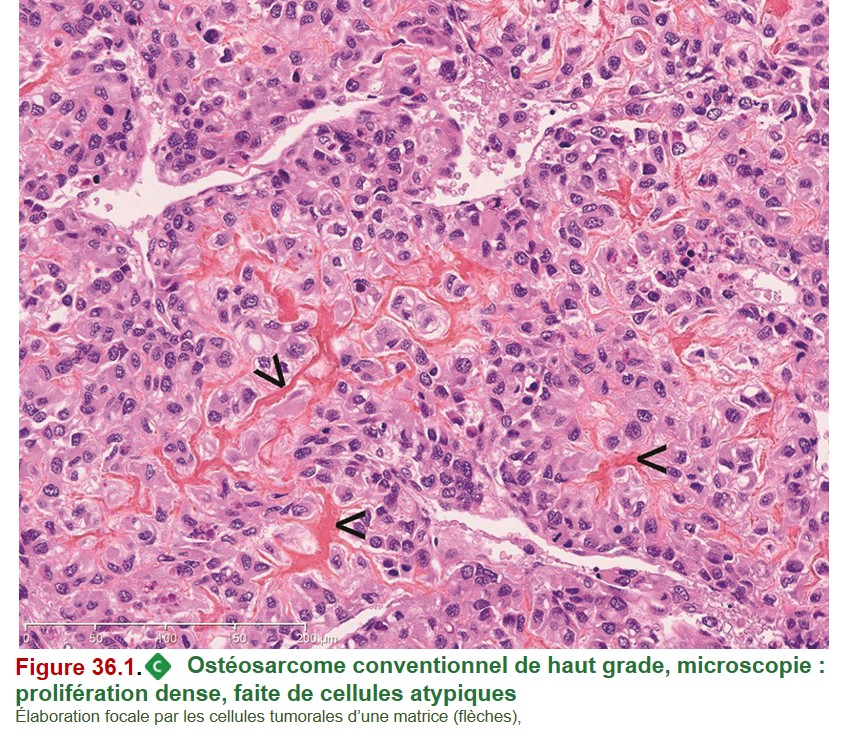
1. Ostéosarcome conventionnel de haut grade
• Histologie : prolifération cellulaire tumorale maligne (ostéoblastes atypiques) élaborant, au moins focalement, une matrice ostéoïde (figure 36.1). À noter que cette tumeur peut produire du cartilage malin et du tissu fibreux malin.
{kind=link}
• Fémur distal +++, tibia proximal, humérus proximal, bassin.
• Dissémination par voie hématogène : métastases pulmonaires.
• Aspect radiologique ostéolytique, ostéocondensant ou mixte.
• Confirmation histologique indispensable avant traitement.
Le traitement habituel de l’ostéosarcome comporte une chimiothérapie néoadjuvante, puis une résection chirurgicale en bloc de la tumeur (c’est-à-dire avec des marges chirurgicales larges, laissant une couche de tissu sain autour de la pièce de résection), et enfin une chimiothérapie adjuvante qui dépend de la réponse histologique de la tumeur. La nécrose tumorale post-chimiothérapie est un élément indispensable du compte rendu anatomopathologique de la pièce de résection, pour l’évaluation de l’efficacité du traitement chimiothérapeutique (conditionne le choix du traitement chimiothérapique post-résection).
2. Sarcome d’Ewing
Cf. Item 299
3. Chondrosarcome central (tumeur cartilagineuse maligne)
• Nodules cartilagineux avec augmentation du nombre des chondrocytes par rapport à un chondrome, atypies cytonucléaires des chondrocytes et critères architecturaux de malignité +++ : résorption active de l’os préexistant par les nodules cartilagineux (diagnostic parfois difficile entre chondrome et chondrosarcome) (figure 36.2).
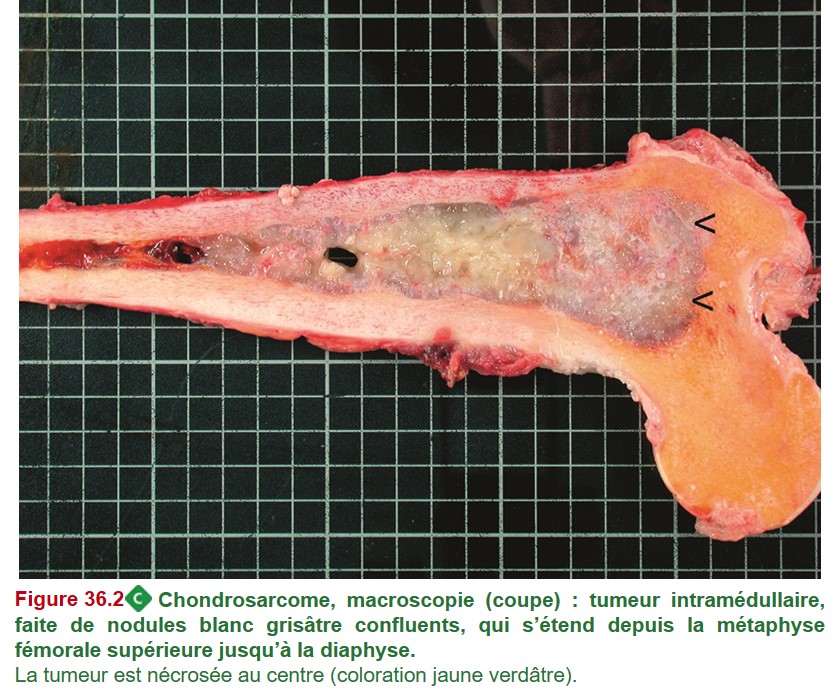{kind=link}
• De novo ou développé à partir d’une lésion cartilagineuse sur le même site (chondrome).
• Diagnostic suspecté dès l’imagerie : tumeur cartilagineuse avec signes d’agression de l’os préexistants (encoches corticales profondes et étendues ± rupture de la corticale et envahissement des tissus mous).
• Preuve histologique nécessaire avant résection.
• Traitement uniquement chirurgical (résection complète en monobloc) car il s’agit d’une tumeur ni chimiosensible, ni radiosensible.
Points clés
• On distingue les tumeurs osseuses primitives (qui peuvent être bénignes ou malignes) des tumeurs secondaires qui sont par définition toujours malignes (métastases par voie hématogène).
• Une biopsie osseuse avec examen anatomopathologique est nécessaire pour établir le diagnostic d’une tumeur osseuse, sauf pour les métastases osseuses d’un cancer primitif connu évolutif ou pour les tumeurs bénignes d’aspect radiologique typique.
• Les biopsies de tumeurs de l’enfant et/ou avec suspicion de sarcome doivent faire l’objet d’une cryopréservation d’un fragment de tumeur non fixé pour éventuelles études moléculaires ultérieures ou bien d’une fixation en formol tamponné sans décalcification.
• La biopsie d’une lésion osseuse suspectée de correspondre à une tumeur osseuse primitive non hématopoïétique doit être faite dans un centre spécialisé (réseau INCa ResOs), car ce sont des tumeurs rares dont la prise en charge diagnostique et thérapeutique nécessite une équipe multidisciplinaire réunissant radiologues, chirurgiens et anatomopathologistes expérimentés.
• La biopsie d’une métastase osseuse permet :
– le diagnostic histologique de la tumeur ;
– l’orientation vers un primitif (pas toujours) ;
– l’identification éventuelle de cibles thérapeutiques.