
CHAPITRE V – Néphrologie
Plan :
• Item 261 –Néphropathie glomérulaire
• Atteinte rénale au cours du myélome
• Item 263 – Néphropathies vasculaires
• Item 195 – Artérite à cellules géantes
• Item 193 – Connaître les principaux types de vascularite systémique, les organes cibles, les outils diagnostiques et les moyens thérapeutiques
Item 261 – Néphropathie glomérulaire
Remarque : ce chapitre aborde également des notions des items 247 – Diabète sucré de types 1 et 2 de l’adulte et de l’enfant – Complications et 194 – Lupus systémique. Syndrome des antiphospholipides (SAPL)
Auteure : Aurélie Sannier
I. Prérequis
II. Introduction
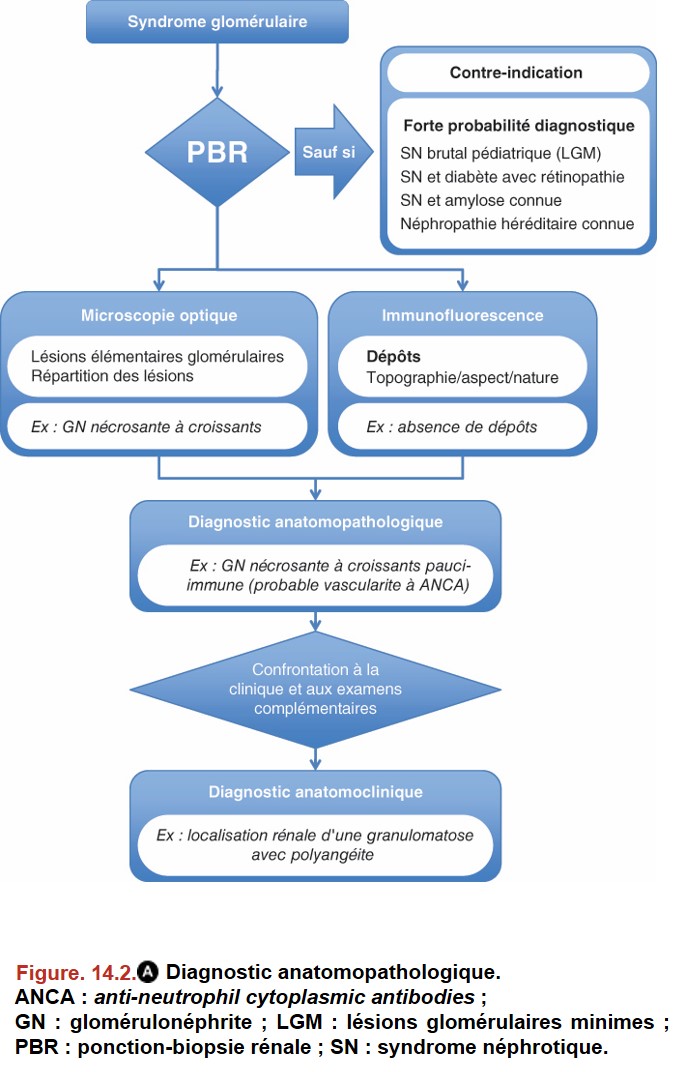
III. Indications de la biopsie rénale
IV. Prise en charge technique anatomopathologique des biopsies rénales
V. Lésions élémentaires glomérulaires
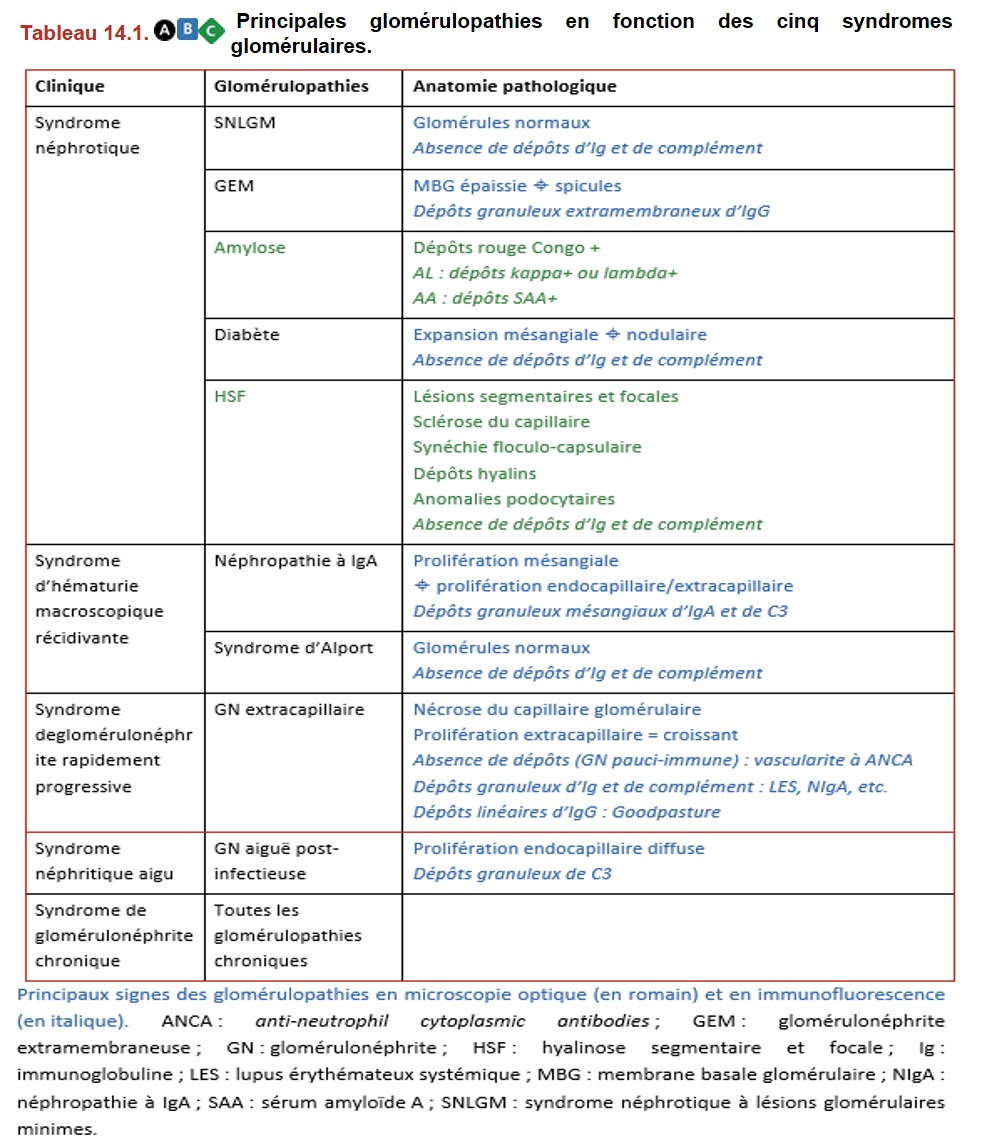
VI. Principales glomérulopathies classées selon leur mode de présentation clinique
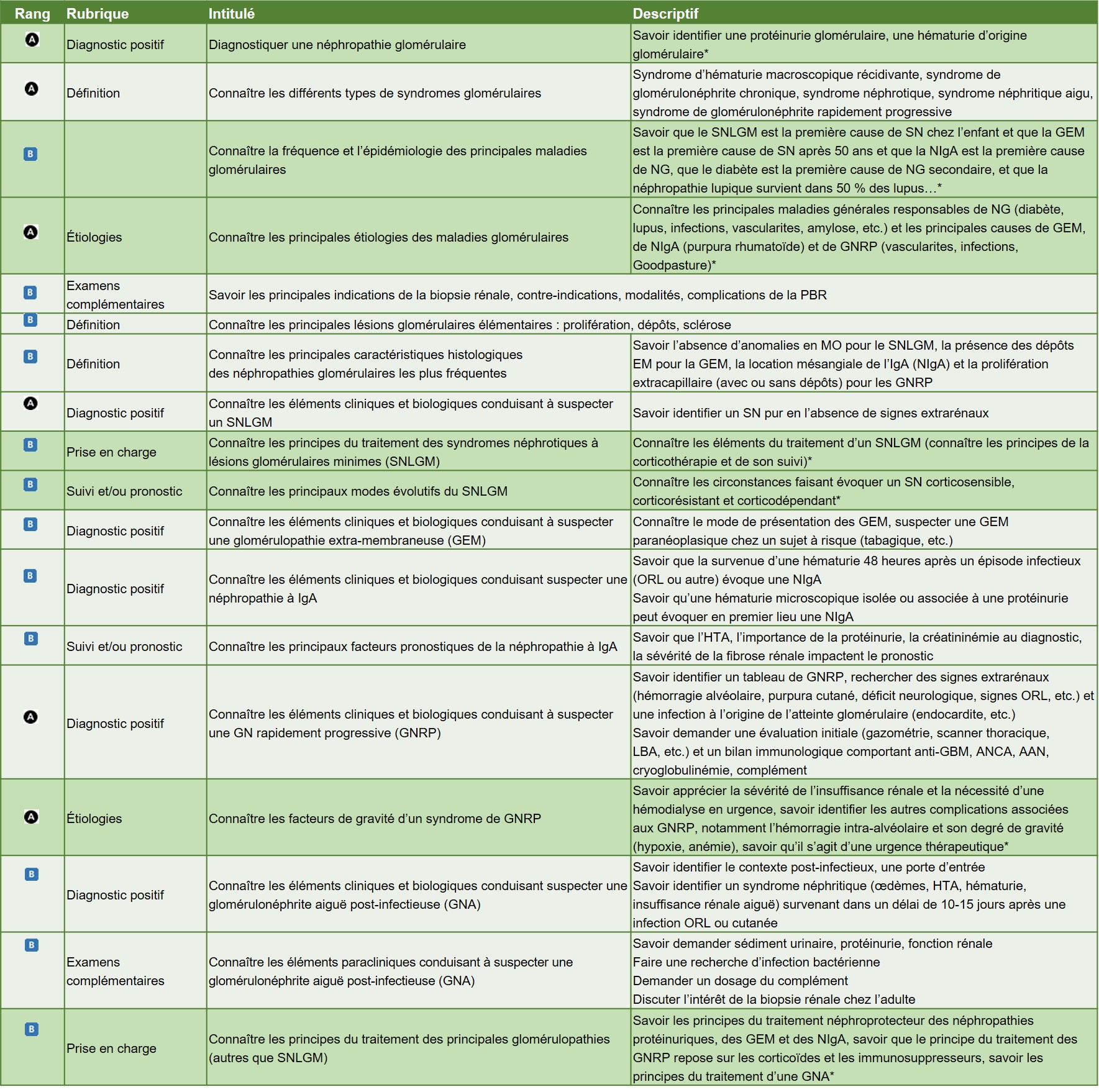
Hiérarchisation des connaissances – Tableau 1
{kind=link}
I. Prérequis
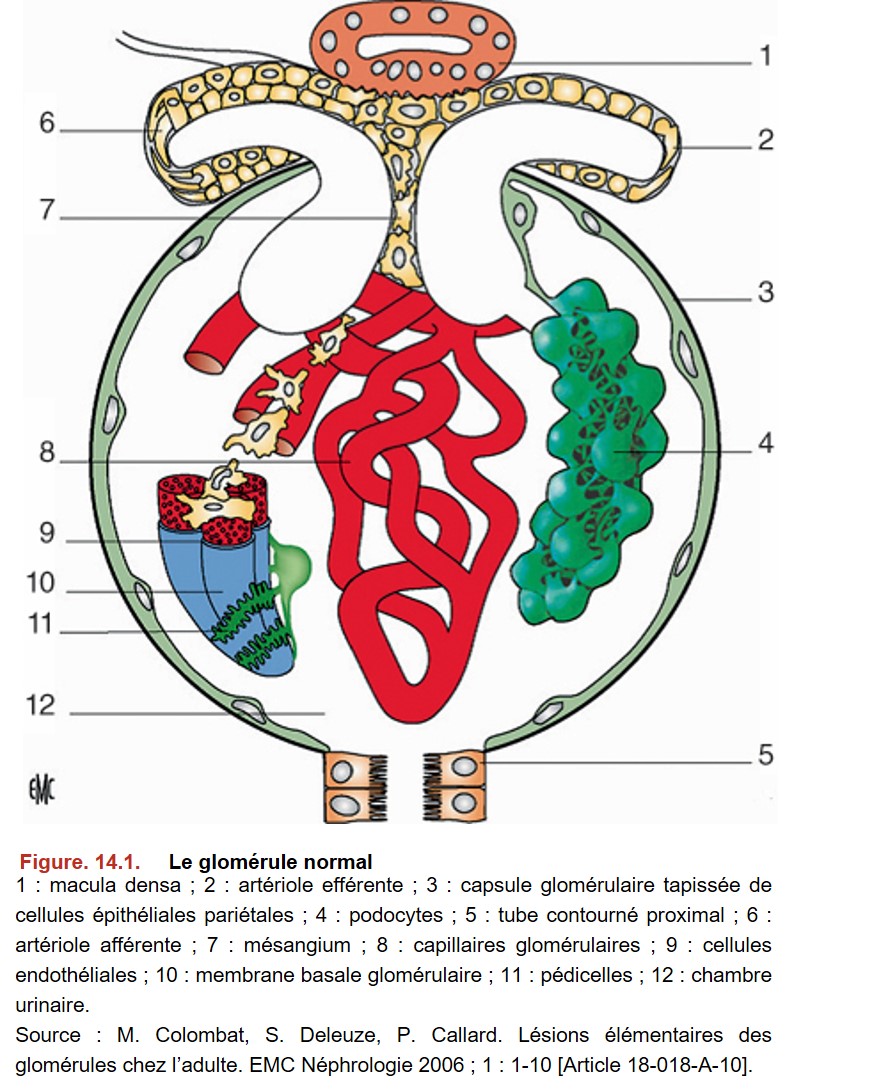
L’histologie glomérulaire est relativement complexe, mais il est indispensable de bien connaître la structure normale du glomérule pour comprendre les principes de la classification anatomopathologique des glomérulopathies (Figure. 14.1).
{kind=link}
La membrane basale glomérulaire (MBG) du capillaire délimite deux espaces :
• l’un situé en dedans de la MBG, appelé espace endocapillaire ou endomembraneux (compartiment sanguin du glomérule) ;
• l’autre situé en dehors de la MBG, appelé espace extracapillaire ou extramembraneux (compartiment urinaire du glomérule).
L’espace endocapillaire comprend les anses capillaires glomérulaires, revêtues de cellules endothéliales. Le mésangium est constitué d’une matrice conjonctive et de cellules (3 à 5 cellules par tige mésangiale). L’espace extracapillaire comprend les podocytes qui recouvrent les anses capillaires, la chambre urinaire, les cellules épithéliales pariétales et la capsule glomérulaire. La MBG se situe entre les podocytes et les cellules endothéliales, ainsi qu’entre les podocytes et le mésangium.
II. Introduction
A. Terminologie
B. Raisonnement médical
L’analyse d’une PBR (ponction-biopsie rénale) aboutit à un diagnostic anatomopathologique. Pour déterminer la maladie en cause (entité anatomoclinique), il est le plus souvent nécessaire de confronter les données anatomopathologiques aux données cliniques et biologiques. Cette confrontation anatomoclinique est une étape fondamentale qui permet en particulier de distinguer les glomérulopathies dites primitives (atteintes glomérulaires sans manifestation extrarénale, le plus souvent idiopathiques) des glomérulopathies dites secondaires qui s’intègrent dans le cadre de maladies générales.
Exemple :
{kind=link}
{kind=link}
III. Indications de la biopsie rénale
• syndrome néphrotique pur chez un enfant âgé de 1 à 10 ans = diagnostic de syndrome à lésions glomérulaires minimes par argument de fréquence ;
• diabète connu avec rétinopathie diabétique au fond d’œil et sans hématurie = diagnostic de glomérulopathie diabétique ;
• amylose documentée sur une biopsie non rénale (glandes salivaires par exemple) = diagnostic de glomérulopathie amyloïde ;
• glomérulopathie héréditaire déjà documentée (par PBR et analyse génétique) dans la famille du patient.
L’apport de la PBR est :
• diagnostique car elle oriente l’enquête étiologique ;
• thérapeutique car elle oriente le traitement de certaines néphropathies glomérulaires ;
• pronostique car elle précise la sévérité des lésions, en particulier pour les lésions dites « chroniques » qui sont irréversibles : proportion de glomérules détruits en « pains à cacheter » et fibrose interstitielle.
IV. Prise en charge technique anatomopathologique des biopsies rénales
La prise en charge technique des PBR au sein des services d’anatomie pathologique conditionne la réalisation du geste lui-même, puisqu’il est nécessaire d’obtenir deux carottes biopsiques : l’une pour l’analyse en microscopie optique et l’autre pour l’immunofluorescence. Un prélèvement supplémentaire est parfois réalisé pour une étude par microscopie électronique (suspicion de syndrome d’Alport, de maladie de dépôts d’immunoglobulines).
• Étude conventionnelle, dite en microscopie optique (MO) :
– à partir du tissu fixé (par le formol ou équivalent) et inclus en paraffine ;
– mise en évidence de lésions élémentaires glomérulaires ;
–
– nombreux plans de coupe pour mise en évidence de lésions segmentaires et/ou focales.
•
– à partir du tissu congelé (prélèvement adressé à « l’état frais » = non fixé) ;
– mise en évidence de dépôts ;
– un même panel d’anticorps est testé sur toute biopsie rénale :
– chaînes lourdes d’immunoglobulines (A, G, M),
– chaînes légères d’immunoglobulines (kappa, lambda),
– fractions du complément (C3, C1q),
– fibrinogène ;
– apporte souvent des informations sur l’étiologie.
MO et IF sont complémentaires et sont effectuées pour toute PBR.
L’étude par microscopie électronique n’est réalisée que dans de rares situations.
V. Lésions élémentaires glomérulaires
A. Principales lésions élémentaires glomérulaires en microscopie optique
1. Proliférations cellulaires
• Prolifération mésangiale : augmentation du nombre de cellules mésangiales.
• Prolifération endocapillaire : augmentation du nombre de cellules du compartiment endocapillaire (situé en dedans de la MBG) : cellules inflammatoires du sang circulant (monocytes, polynucléaires neutrophiles), cellules endothéliales. Elle reflète une inflammation glomérulaire et est toujours la conséquence de la présence de dépôts endomembraneux.
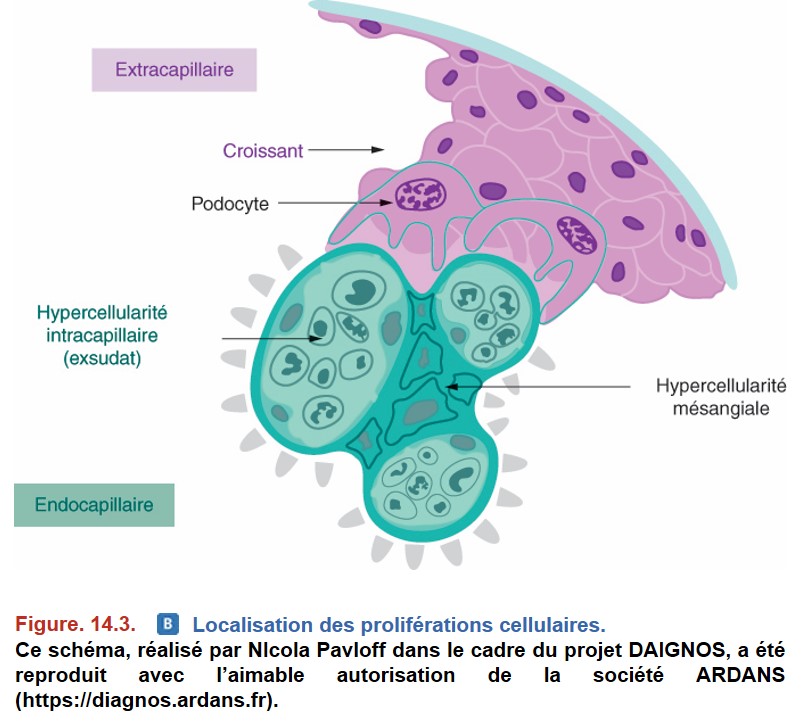
• Prolifération extracapillaire : augmentation du nombre de cellules épithéliales pariétales à la suite d’une nécrose de la paroi du capillaire glomérulaire :
– formation d’un « croissant » dans la chambre urinaire (le terme de croissant provient de la forme prise par la lésion sur une coupe histologique en deux dimensions) ;
– évolution cicatricielle vers la fibrose (« organisation » du croissant) (Figure. 14.3).
{kind=link}
Le terme d’« hypercellularité » est parfois utilisé comme synonyme de « prolifération ».
2. Sclérose (ou fibrose)
Il s’agit de l’accumulation cicatricielle d’un matériel de nature collagénique remplaçant la totalité (glomérule scléreux ou « pain à cacheter ») ou une partie (sclérose segmentaire) du glomérule lésé. Une augmentation de la matrice conjonctive du mésangium est également visible dans le diabète.
3. Répartition des lésions
• Lésions focales : ne touchent qu’une minorité des glomérules. À l’inverse, les lésions diffuses touchent tous les glomérules de la même manière.
• Lésions segmentaires : pour un glomérule donné, seule une partie du glomérule est atteinte. À l’inverse, les lésions globales touchent la totalité du glomérule atteint.
B. Analyse des dépôts (IF)
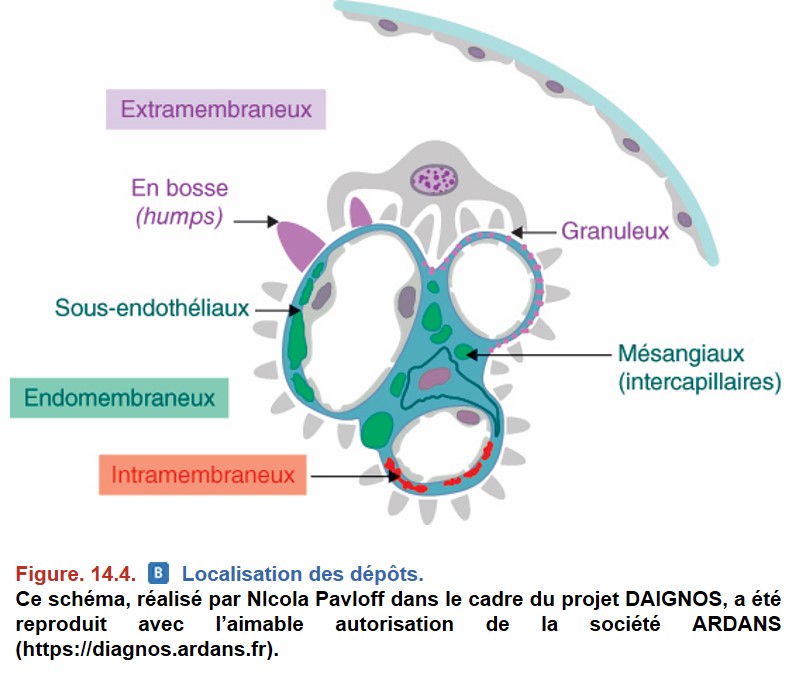
1. Localisation des dépôts
• Dépôts mésangiaux : dépôts situés au sein du mésangium.
• Dépôts extramembraneux : dépôts immuns situés sur le versant externe de la MBG (entre la MBG et le podocyte).
• Dépôts endomembraneux : dépôts immuns situés en dedans de la MBG (versant interne) dans l’espace sous-endothélial (entre la cellule endothéliale et la MBG).
• Dépôts intramembraneux, situés dans la membrane basale : beaucoup plus rares (Figure. 14.4).
{kind=link}
2. Aspect des dépôts en immunofluorescence
• Dépôts granuleux, de loin les plus fréquents, correspondant aux dépôts « immuns » constitués d’Ig (immunoglobulines) et de complément (GEM [glomérulonéphrite extramembraneuse], IgA, lupus, cryoglobulinémie, GNA [glomérulonéphrite aiguë] post-streptococcique).
• Dépôts linéaires, beaucoup plus rares (glomérulonéphrites à anticorps anti-MBG, certaines formes de maladies à dépôts d’immunoglobulines monoclonales).
• Dépôts plus volumineux et homogènes :
– dépôts « hyalins » ne correspondant pas à des dépôts immuns (hyalinose segmentaire et focale [HSF] primitive) ;
– amylose.
3. Nature des dépôts en immunofluorescence
• Immunoglobulines (IgA, IgG, IgM, kappa, lambda).
• Fractions du complément (C1q, C3).
Remarque :
Remarque :
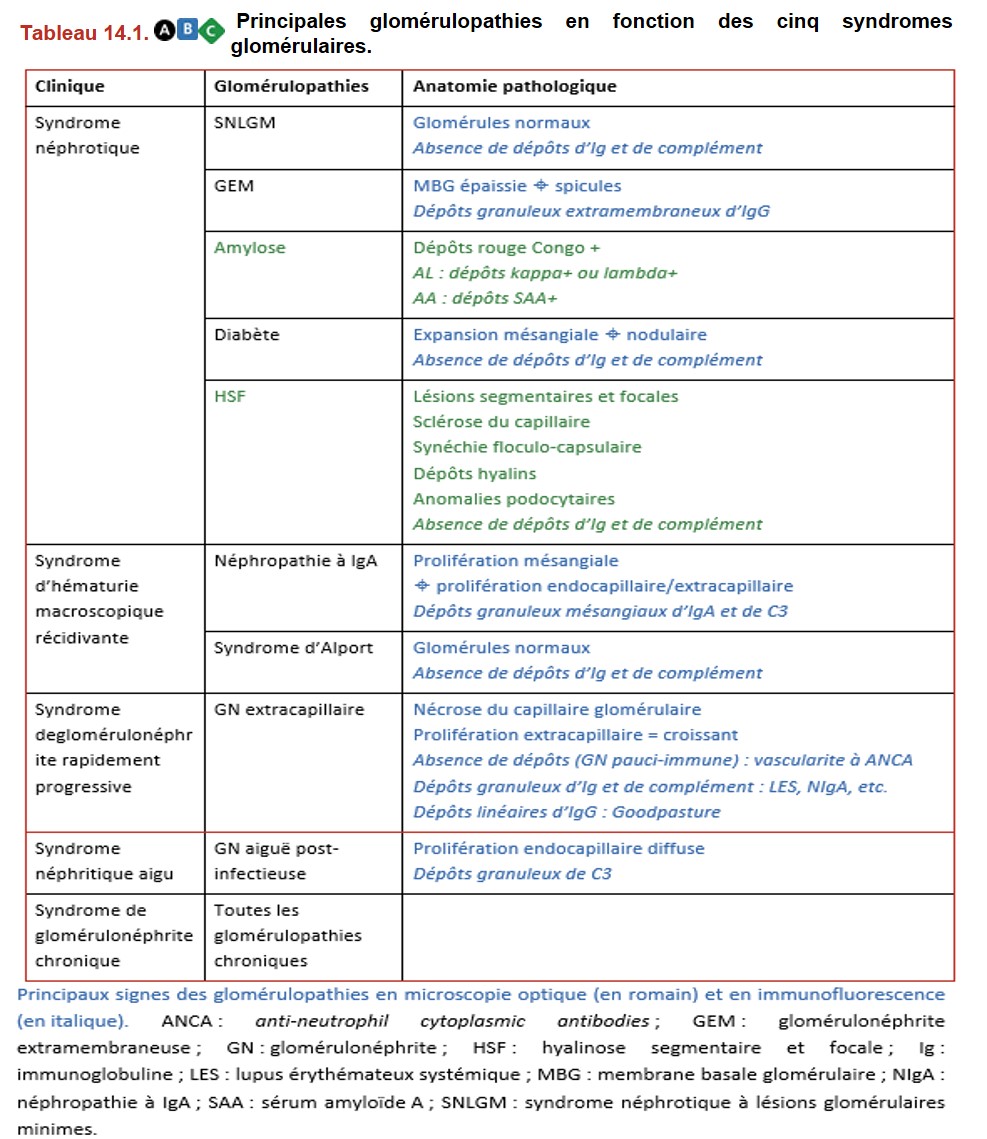
VI. Principales glomérulopathies classées selon leur mode de présentation clinique
{kind=link}
A. Syndrome néphrotique
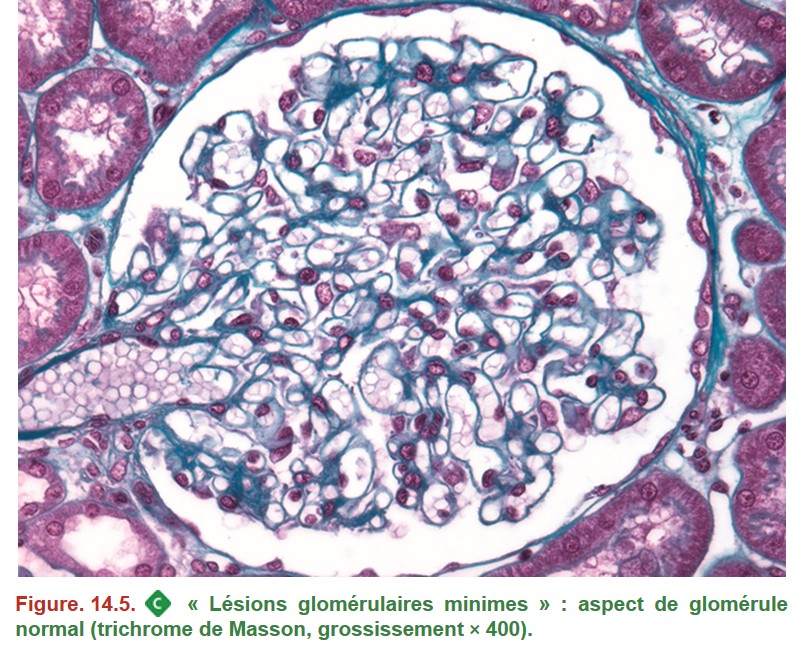
1. Syndrome néphrotique à lésions glomérulaires minimes (SNLGM)
•
• IF : absence de dépôts.
• Microscopie électronique : fusion des pédicelles (pieds des podocytes).
Chez l’enfant entre 1 et 10 ans, lorsque la clinique est typique (syndrome néphrotique pur), il s’agit de l’une des rares situations où la PBR n’est pas nécessaire pour identifier la glomérulopathie en cause, le diagnostic étant retenu par argument de fréquence.
{kind=link}
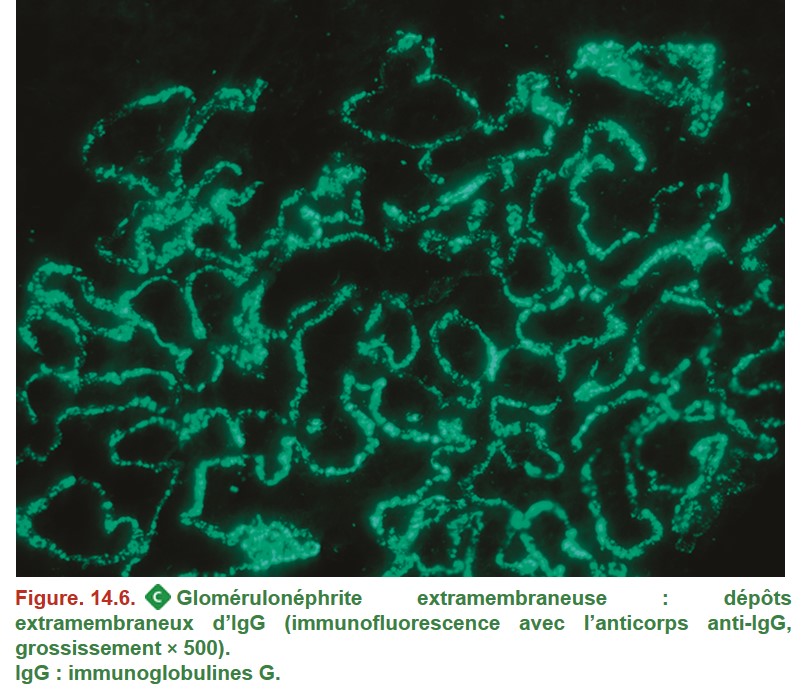
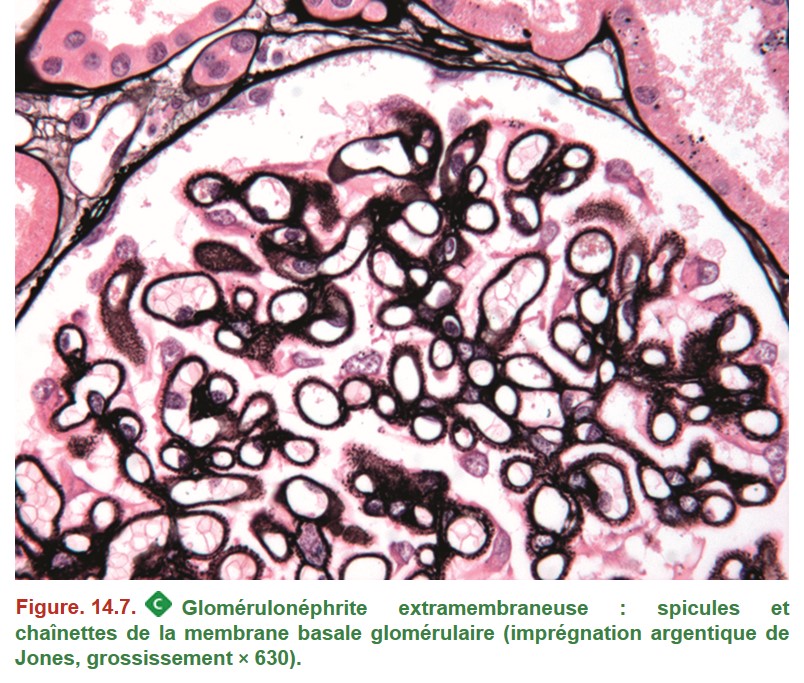
2. Glomérulonéphrite extramembraneuse
La GEM est due à la présence de dépôts immuns sur le versant externe de la MBG. Le diagnostic de GEM peut être porté sans biopsie rénale en cas de détection d’anticorps sériques dirigés contre le récepteur de la phospholipase A2 (anti-PLA2R) chez un patient ayant un syndrome néphrotique.
• MO : MBG normale ou épaissie et spiculée sur son versant externe ;
• IF (Figure. 14.6) :
{kind=link}
– dépôts granuleux extramembraneux ;
– constitués d’IgG et de C3.
{kind=link}
En cas de GEM liée à une immunisation anti-PLA2R, la mise en évidence de l’antigène PLA2R accumulé au sein des dépôts extramembraneux peut être faite par immunohistochimie sur la PBR. D’autres antigènes cibles ont été identifiés ces dernières années dans les GEM PLA2R-négatives ; ces antigènes peuvent également être détectés par immunohistochimie.
3. Amylose
Les amyloses sont un ensemble de maladies caractérisées par des dépôts tissulaires extracellulaires constitués de protéines insolubles, fibrillaires, organisées en feuillets bêta plissés. Le diagnostic nécessite la mise en évidence anatomopathologique de ces dépôts. On doit privilégier la réalisation de biopsies peu invasives (biopsie de glandes salivaires accessoires par exemple) avant d’envisager une PBR.
Les amyloses les plus fréquentes au niveau du rein sont :
• l’amylose AL, dérivée de chaînes légères d’immunoglobulines, lors d’une prolifération tumorale plasmocytaire (MGUS [monoclonal gammopathy of undetermined significance] plus souvent que myélome) ;
• l’amylose AA, dérivée de la protéine sérum amyloïde A (SAA), compliquant des maladies inflammatoires chroniques : infections prolongées, polyarthrite rhumatoïde, maladies inflammatoires chroniques de l’intestin, fièvre méditerranéenne familiale, etc.
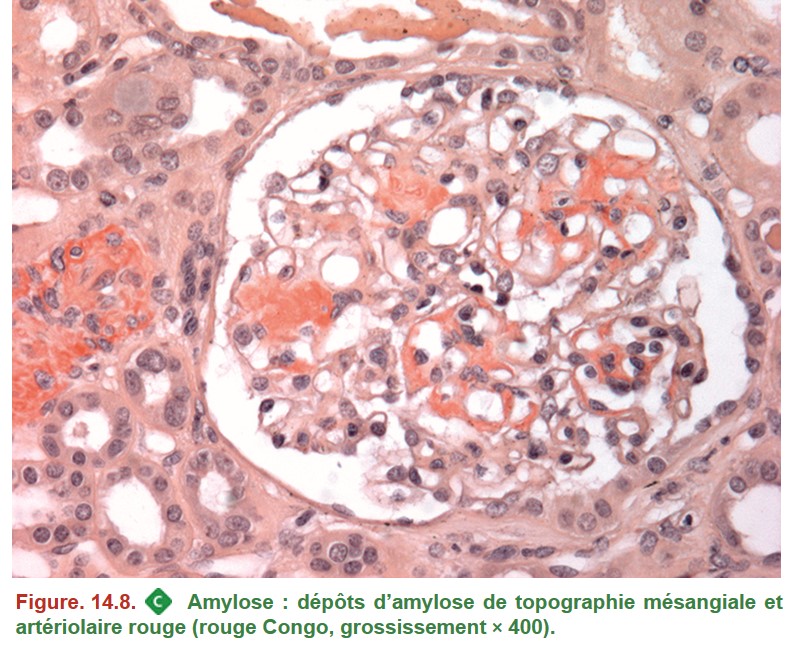
Microscopie optique
• Dépôts extracellulaires amorphes, homogènes, éosinophiles à l’HE ou HES.
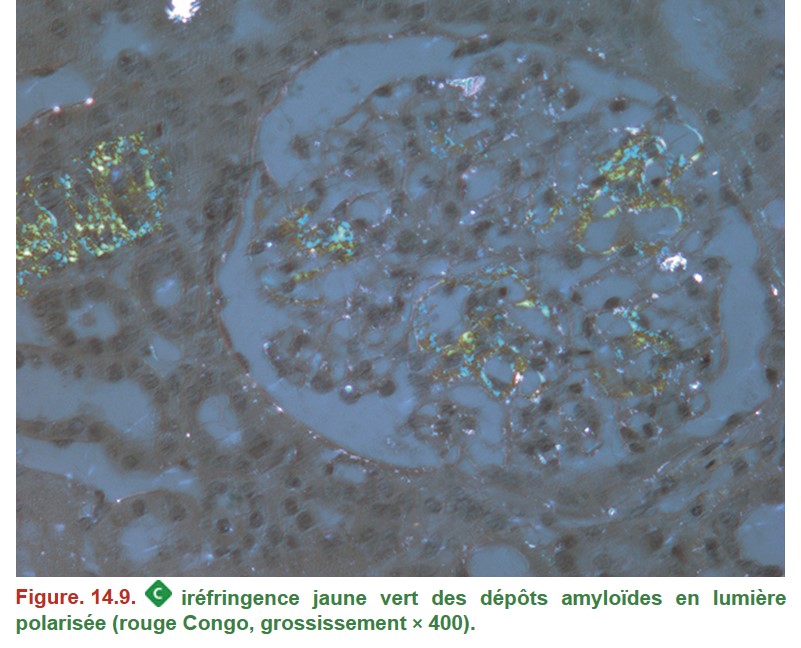
• Rouge Congo :
– coloration spécifique de l’amylose, indispensable au diagnostic (Figure. 14.8) ;
{kind=link}
– dépôts « congophiles » avec biréfringence jaune vert en lumière polarisée (Figure. 14.9).
{kind=link}
• Les dépôts ne se limitent pas aux glomérules et peuvent toucher tous les constituants du tissu rénal :
– glomérules : mésangium, parois capillaires ;
– parois des vaisseaux ;
– interstitium.
IF et immunohistochimie (pour typage de l’amylose)
• Amylose AL : dépôts monotypiques de chaînes légères d’immunoglobulines, c’est-à-dire marqués par l’anticorps anti-kappa ou anti-lambda (en fonction de la chaîne légère en cause).
• Amylose AA : marquage des dépôts par l’anticorps anti-SAA (technique réalisable en immunohistochimie sur tissu fixé et inclus en paraffine).
4. Diabète
• MO :
– augmentation de la taille des glomérules (hypertrophie glomérulaire) ;
– puis augmentation de la matrice mésangiale du fait de l’accumulation de matrice extracellulaire glycosylée (expansion mésangiale diffuse) ;
– puis expansion mésangiale nodulaire à un stade plus évolué (« nodules de Kimmelstiel-Wilson ») avec épaississement de la MBG et dépôts hyalins glomérulaires ;
– au stade terminal, sclérose glomérulaire globale et fibrose interstitielle étendue ;
– lésions vasculaires associées : hyalinose artériolaire.
• IF : absence de dépôts de type immun.
Lorsqu’un syndrome de néphropathie glomérulaire survient progressivement chez un patient ayant un diabète connu avec rétinopathie diabétique documentée, un diagnostic présomptif de néphropathie diabétique est retenu et la PBR n’est pas réalisée.
5. Hyalinose segmentaire et focale primitive
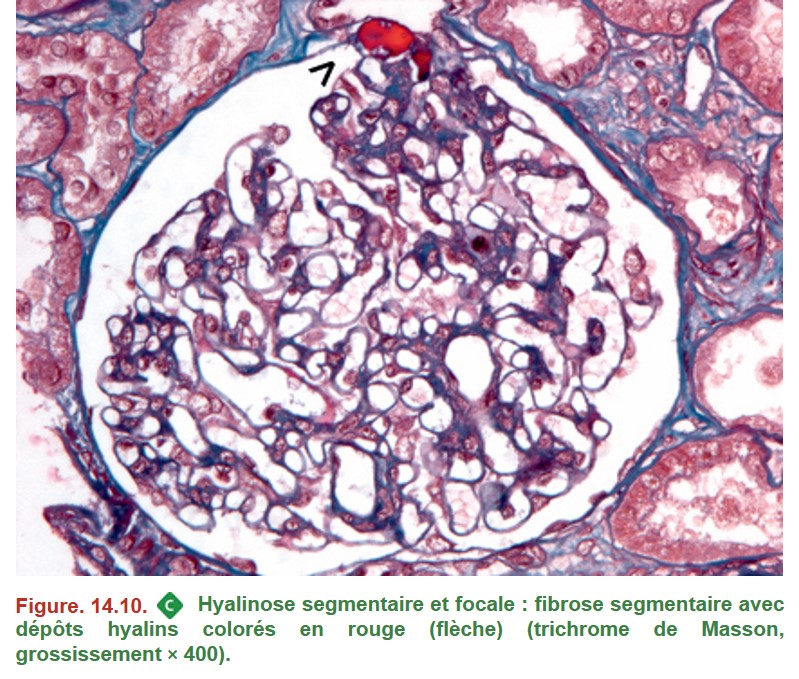{kind=link}
• MO :
– lésions segmentaires et focales ;
– sclérose : fibrose mésangiale entraînant une oblitération du capillaire glomérulaire ;
– synéchie floculo-capsulaire : adhérence entre le capillaire glomérulaire et la capsule glomérulaire ;
– volumineux dépôts hyalins (protéines plasmatiques « piégées » dans les zones de fibrose) ;
– anomalies podocytaires : vacuolisation, hypertrophie et hyperplasie des podocytes.
• IF :
– absence de dépôts de type immun ;
– fixation non spécifique au niveau des dépôts hyalins (IgM, C3).
B. Syndrome d’hématurie macroscopique récidivante
1. Néphropathie à IgA
• MO :
– épaississement mésangial prolifération mésangiale ;
– prolifération endocapillaire extracapillaire ;
– sclérose glomérulaire (poussées antérieures) ;
–
•
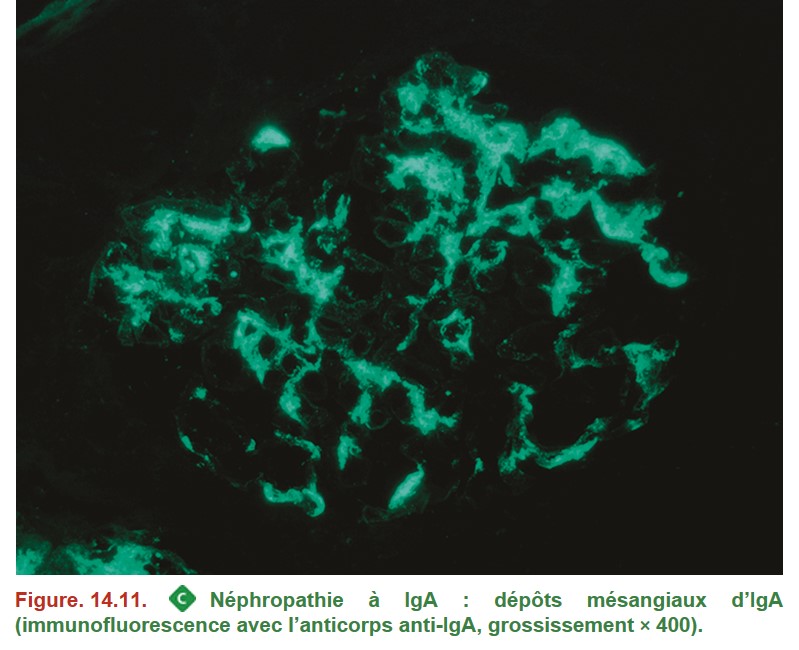{kind=link}
2. Syndrome d’Alport
•
• IF :
– absence de dépôts ;
– étude de la distribution des chaînes alpha du collagène IV à l’aide d’anticorps spécifiques : perte d’expression de la chaîne alpha-5 du collagène IV au niveau des membranes basales glomérulaires et tubulaires. L’examen en microscopie électronique de la biopsie rénale peut également mettre en évidence des anomalies de la membrane basale glomérulaire évocatrices du diagnostic.
C. Syndrome de glomérulonéphrite rapidement progressive (GNRP)
1. Données communes à l’ensemble des syndromes de GNRP
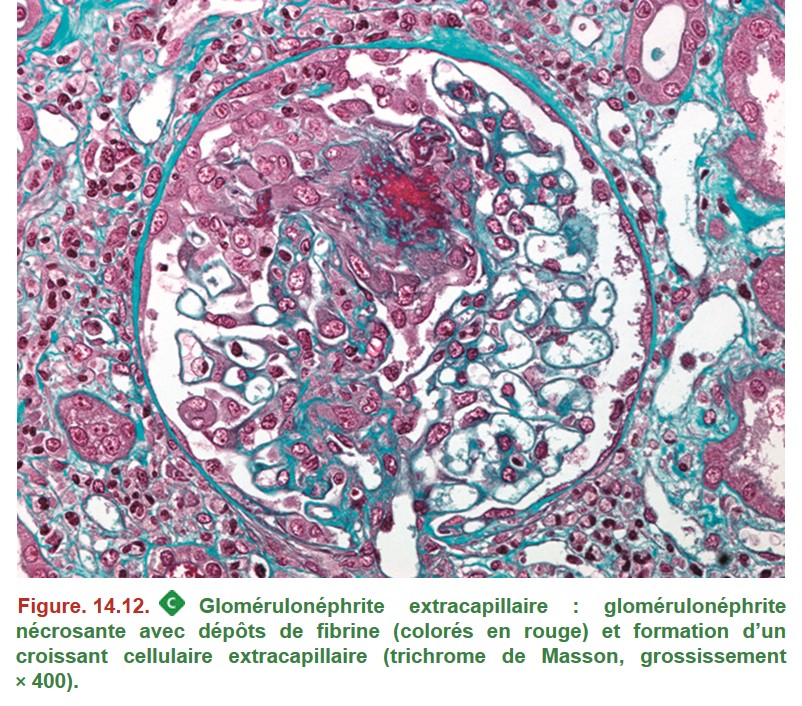{kind=link}
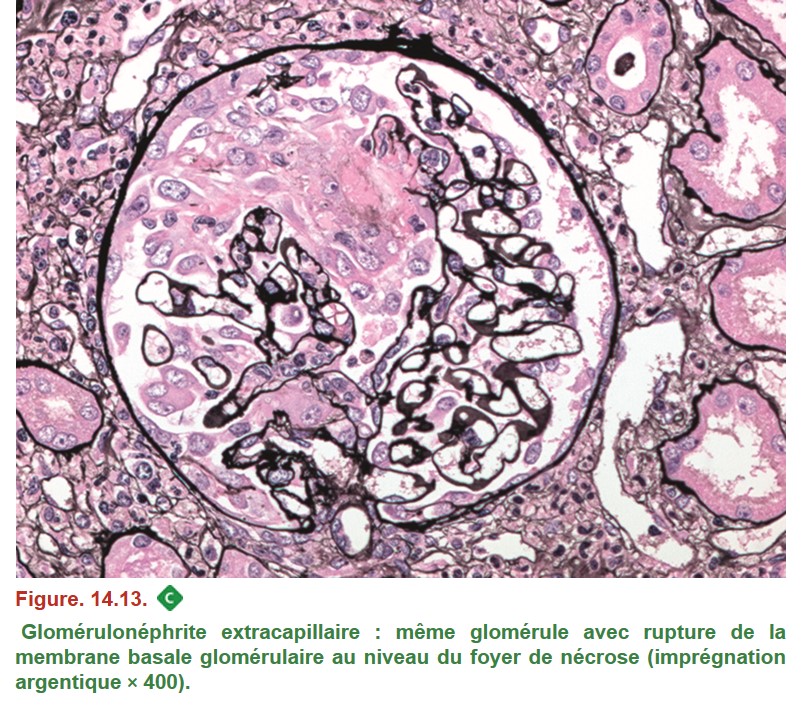{kind=link}
•
– foyers de nécrose de la paroi des capillaires glomérulaires avec dépôts de fibrine ;
– prolifération extracapillaire = croissant formé à partir des cellules épithéliales pariétales ;
– prolifération endocapillaire associée si présence de dépôts immuns endomembraneux (lupus, IgA, cryoglobulinémie) ;
–
•
– absence de dépôt (≈ 60 % des cas) :
– vascularite dite pauci-immune (car absence de dépôts d’Ig en IF) à ANCA,
– étiologies : granulomatose avec polyangéite (anciennement maladie de Wegener), polyangéite microscopique ;
– dépôts immuns endomembraneux granuleux d’Ig et/ou de complément (≈ 30 %) :
– prolifération endocapillaire associée,
– étiologies : lupus (dépôts d’IgG, d’IgA, d’IgM, de C3 et de C1q), néphropathie à IgA (dépôts d’IgA et de C3), cryoglobulinémie (dépôts d’IgG, d’IgM, de C3 ± de C1q), glomérulonéphrite aiguë post-infectieuse (dépôts de nature variable en fonction de l’agent pathogène responsable, souvent à prédominance de C3) ;
– dépôts linéaires d’IgG le long de la MBG (≈ 10 %) : maladie de Goodpasture.
2. La GNRP, un des modes de presentation de la néphropathie lupique
Remarque :
L’atteinte rénale doit être recherchée (bandelette urinaire ou évaluation du rapport protéinurie/créatininurie) régulièrement au cours du suivi d’une maladie lupique. Une PBR est indiquée devant une protéinurie supérieure ou égale à 0,5 g/j (ou 0,5 g/g de créatininurie) et/ou en cas d’insuffisance rénale. La grande hétérogénéité des formes d’atteinte glomérulaire du lupus conditionne le mode de présentation clinique néphrologique, qui est donc très variable : hématurie microscopique, protéinurie glomérulaire ± syndrome néphrotique, syndrome de GNRP, insuffisance rénale de degré variable.
•
– glomérulonéphrite proliférative endocapillaire ;
– glomérulonéphrite nécrosante à croissants ;
– glomérulonéphrite extramembraneuse.
• IF : dépôts d’immunoglobulines d’IgA, d’IgG, d’IgM, de C1q et de C3.
{kind=link}
D. Syndrome néphritique
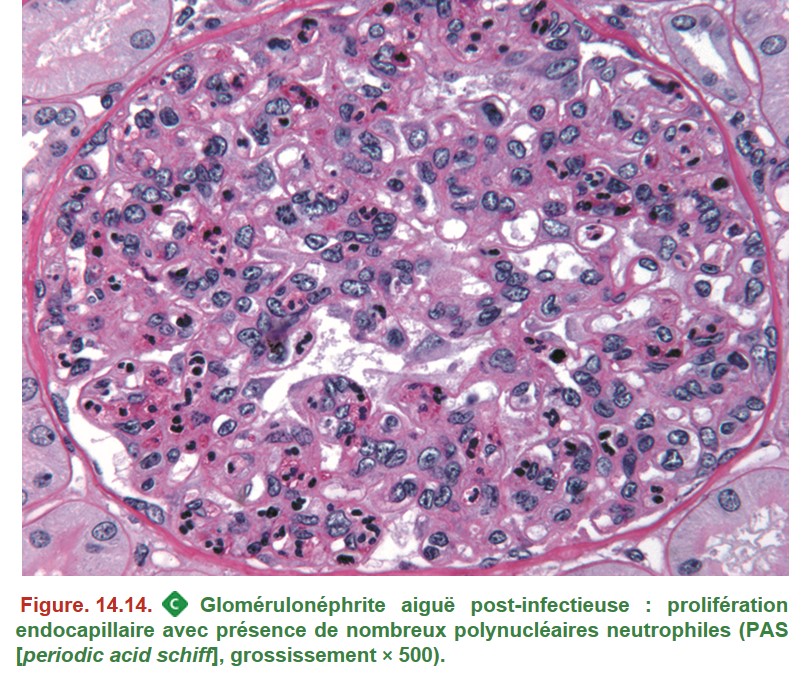
C’est le mode de présentation habituel des GNA post-infectieuses (classiquement post-streptococciques). La biopsie rénale est réalisée en cas de doute diagnostique.
• MO (Figure. 14.14) :
{kind=link}
– prolifération endocapillaire diffuse : afflux de cellules inflammatoires circulantes (polynucléaires neutrophiles ++) ;
– volumineux dépôts extramembraneux irrégulièrement répartis (humps) ;
– prolifération extracapillaire possible dans les formes sévères.
• IF : dépôts immuns mésangiaux et dans la paroi des capillaires glomérulaires (aspect en « ciel étoilé ») ; majoritairement constitués de C3.
Points clés
• Il est indispensable de bien connaître la structure normale du glomérule pour comprendre les principes de la classification anatomopathologique des glomérulopathies ainsi que la terminologie utilisée.
• La biopsie rénale est l’examen de référence pour le diagnostic des néphropathies glomérulaires.
• Un examen en microscopie conventionnelle (dite « optique ») et un examen en immunofluorescence doivent être réalisés, d’où la nécessité de faire deux biopsies, l’une qui est fixée et l’autre congelée.
• L’examen en microscopie optique recueille des lésions élémentaires, tandis que l’examen en immunofluorescence met en évidence des dépôts.
• Le diagnostic est souvent anatomoclinique par confrontation des lésions anatomopathologiques et des données cliniques.
Atteinte rénale au cours du myélome
Remarque : chapitre rattaché à l’item 320 – Myélome multiple des os, abordé hors atteinte rénale dans le chapitre correspondant à l’item 320.
Auteure : Aurélie Sannier
I. Néphropathie à cylindres myélomateux ou tubulopathie myélomateuse
II. Amylose AL
III. Maladie des dépôts d’immunoglobulines monoclonales (monoclonal immunoglobulin deposition disease, MIDD) ou maladie de Randall
IV. Syndrome de Fanconi associé au myélome
Hiérarchisation des connaissances / Item 320 – Myélome multiple des os / Tableau 2
{kind=link}
Introduction
L’insuffisance rénale est présente chez environ 20 % des patients au moment du diagnostic de myélome. La découverte d’une protéinurie de type glomérulaire avec une albuminurie supérieure à 1 g/j doit faire suspecter une néphropathie glomérulaire associée au myélome telle qu’une amylose AL ou une maladie de dépôts d’immunoglobulines monoclonales de type Randall.
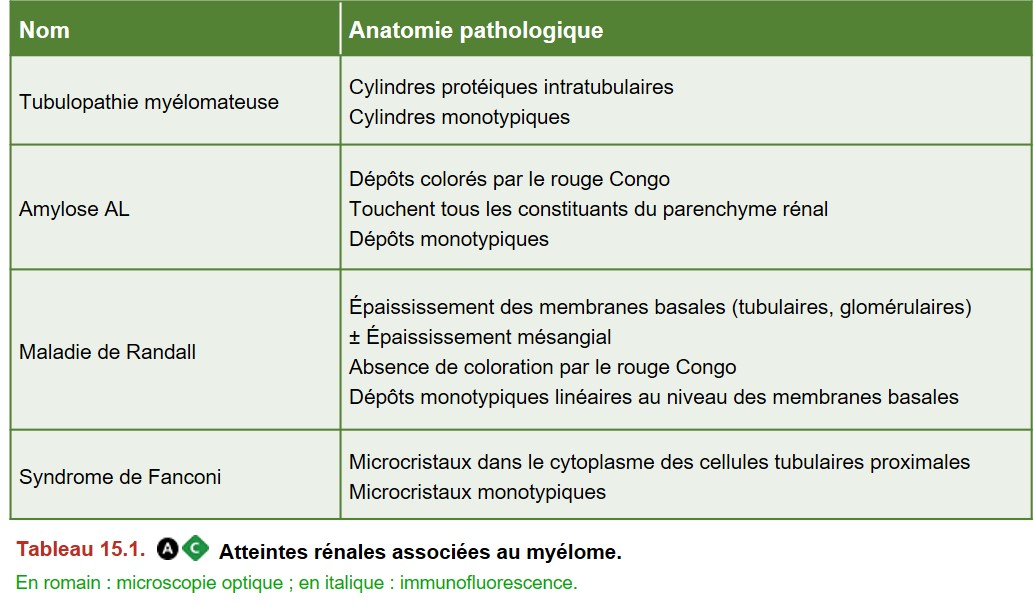
Les atteintes rénales sont principalement (Tableau 15.1) :
{kind=link}
• la néphropathie à cylindres myélomateux (NCM) ou tubulopathie myélomateuse ;
• l’amylose AL ;
• la maladie à dépôts d’immunoglobulines monoclonales (ou maladie de Randall) ;
•
Remarque :
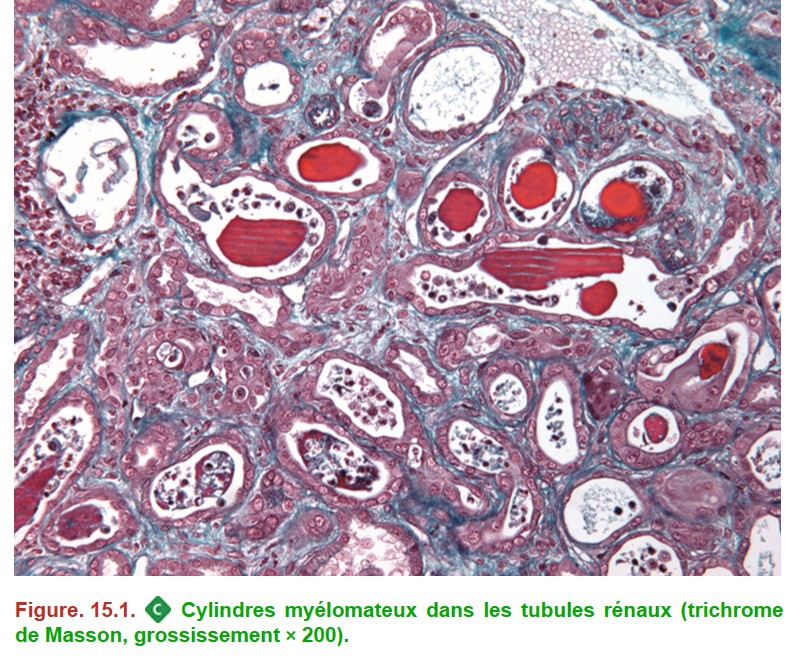
I. Néphropathie à cylindres myélomateux ou tubulopathie myélomateuse
C’est la complication rénale la plus fréquente du myélome, liée à la précipitation intratubulaire de chaînes légères d’immunoglobulines monoclonales entraînant une insuffisance rénale.
La tubulopathie myélomateuse complique les myélomes avec élimination urinaire de chaînes légères.
Le diagnostic peut être porté sans réalisation de biopsie rénale (preuve histologique non indispensable) chez un patient atteint d’un myélome connu et présentant une insuffisance rénale aiguë avec protéinurie constituée majoritairement de chaînes légères, en particulier si un facteur favorisant la précipitation intratubulaire des chaînes légères est identifié.
•
– cylindres protéiques intratubulaires (prédominant au niveau des tubules distaux) polychromatophiles, fracturés, avec cellules géantes au contact ;
– altérations de l’épithélium tubulaire secondaires à la formation des cylindres = nécrose tubulaire aiguë (Figure. 15.1) ;
{kind=link}
• IF : fixation de l’une des deux chaînes légères (kappa ou lambda) au niveau des cylindres protéiques (monotypie des cylindres).
II. Amylose AL
•
– dépôts amorphes, vert pâle au trichrome de Masson, éosinophiles à l’HE ou HES ;
– rouge Congo indispensable pour confirmer le diagnostic (coloration spécifique de l’amylose) (Figure. 15.2) :
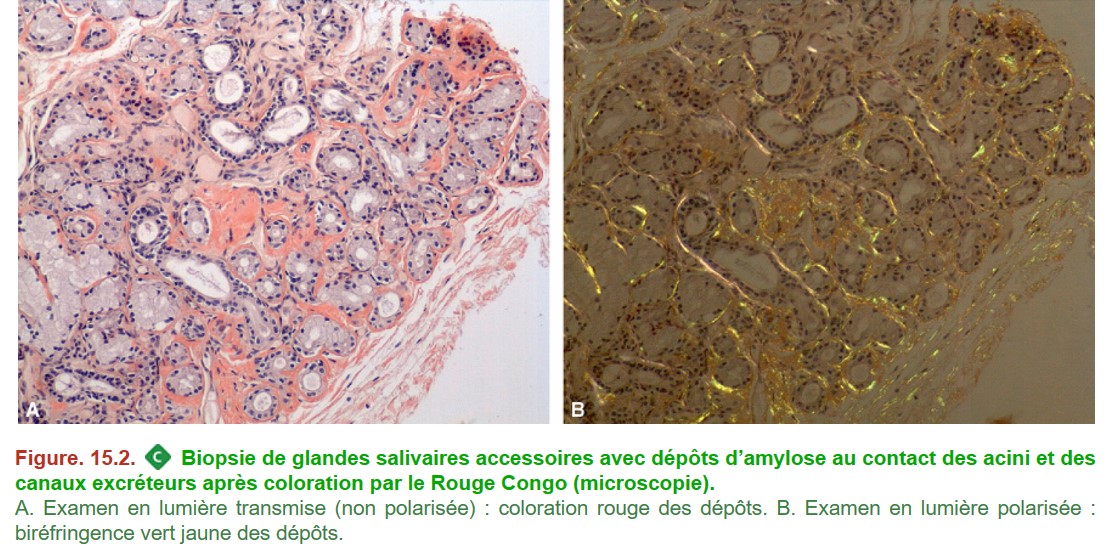{kind=link}
– dépôts rouges en lumière blanche transmise (examen microscopique habituel) (« congophiles »),
– biréfringence vert jaune en lumière polarisée ;
– les dépôts ne se limitent pas aux glomérules et peuvent toucher tous les constituants du tissu rénal :
– glomérules (mésangium, parois capillaires),
– parois des vaisseaux,
– interstitium.
• IF : dépôts (glomérulaires et/ou vasculaires et/ou interstitiels) monotypiques (le plus souvent lambda).
Le diagnostic peut être porté grâce à des prélèvements biopsiques moins invasifs que la PBR, comme la biopsie des glandes salivaires accessoires.
L’amylose AL est une maladie systémique qui peut atteindre, en plus du rein, de nombreux organes : myocarde, nerfs périphériques, etc.
III. Maladie des dépôts d’immunoglobulines monoclonales ou maladie de Randall
• MO :
– dépôts non colorés par le rouge Congo ;
– localisation préférentielle des dépôts au niveau des membranes basales (tubulaires et glomérulaires) qui apparaissent épaissies ;
– ± épaississement mésangial ressemblant à la glomérulosclérose diabétique.
• IF : dépôts linéaires monotypiques (kappa plus souvent que lambda) au niveau des membranes basales tubulaires et glomérulaires ainsi que du mésangium (Figure. 15.3).
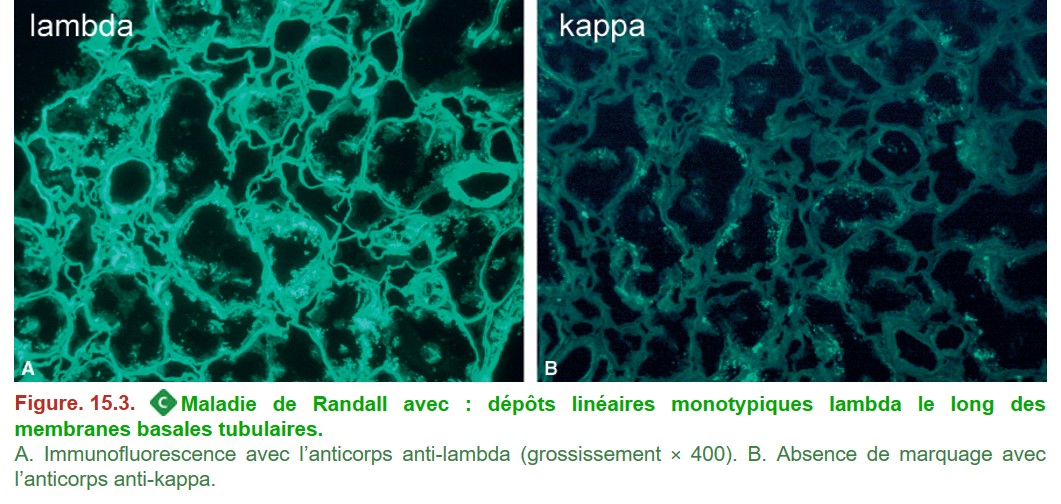{kind=link}
IV. Syndrome de Fanconi associé au myélome
Il est rare et secondaire à un dysfonctionnement tubulaire proximal lié à l’accumulation intracytoplasmique de microcristaux constitués de chaînes légères d’immunoglobulines monotypiques, entraînant un trouble de la réabsorption au niveau des cellules épithéliales tubulaires proximales.
• MO : cellules tubulaires proximales avec microcristaux dans le cytoplasme + altérations épithéliales (vacuolisation cytoplasmique).
• IF : chaînes légères monotypiques (kappa ou lambda) d’immunoglobulines au niveau des cristaux.
Points clés
• L’atteinte rénale au cours du myélome tient une part importante dans le pronostic et la décision thérapeutique.
• Les atteintes rénales sont principalement :
– la néphropathie à cylindres myélomateux ou tubulopathie myélomateuse. C’est la complication rénale la plus fréquente du myélome ; la biopsie rénale n’est pas indispensable pour son diagnostic ;
– une biopsie rénale peut être justifiée pour rechercher une amylose AL ou une maladie à dépôts d’immunoglobulines monoclonales (ou maladie de Randall) en cas de protéinurie glomérulaire.
Item 263 – Néphropathies vasculaires
Auteur : Jean-Baptiste Gibier
I. Introduction
II. Néphroangiosclérose bénigne
III. Microangiopathie thrombotique
IV. Maladies des emboles de cristaux de cholestérol
Hiérarchisation des connaissances : Tableau 3
{kind=link}
I. Introduction
II. Néphroangiosclérose « bénigne »
C’est le retentissement rénal d’une HTA ancienne et insuffisamment contrôlée. Elle entraîne l’apparition d’une insuffisance rénale chronique (IRC) avec une détérioration progressive de la fonction rénale pouvant aboutir à une insuffisance rénale terminale. Il s’agit d’une cause fréquente d’IRC.
C’est un diagnostic d’exclusion et les lésions histologiques sont peu spécifiques. La biopsie rénale peut être réalisée pour ne pas méconnaître une autre néphropathie.
•
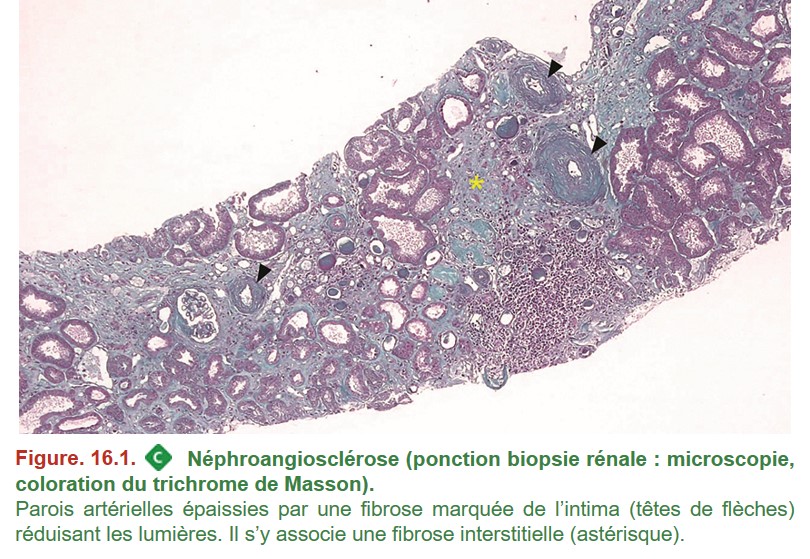{kind=link}
– épaississement fibreux de l’intima des artères ou endartérite fibreuse (artériosclérose) ;
– dépôts hyalins artériolaires (artériolosclérose) ;
– fibrose interstitielle et atrophie tubulaire ;
– signes d’ischémie glomérulaire ;
– prédominance des glomérules scléreux dans la région sous-capsulaire.
• IF : absence de dépôts.
III. Microangiopathie thrombotique
• SHU (syndrome hémolytique et urémique) dit « typique » (post-diarrhéique, surtout chez l’enfant) : infection à entérobactéries productrices de shiga-like toxins : E. coli O157 :H7 ++ ;
• SHU dit « atypique » secondaire à une anomalie de la voie alterne du complément (acquise ou génétique) ;
• purpura thrombotique thrombocytopénique ;
• médicaments (ciclosporine, gemcitabine, quinine, etc.) ;
• MAT compliquant une maladie sous-jacente : HTA maligne, syndrome des antiphospholipides, sclérodermie, cancers, infections ;
• éclampsie.
La biopsie rénale n’est pas systématiquement indiquée, en particulier dans le SHU typique de l’enfant où elle est inutile, mais elle est souvent réalisée chez l’adulte après équilibre tensionnel pour rechercher une néphropathie sous-jacente.
MO – les lésions touchent les artérioles et/ou les glomérules (Figure. 16.2) :
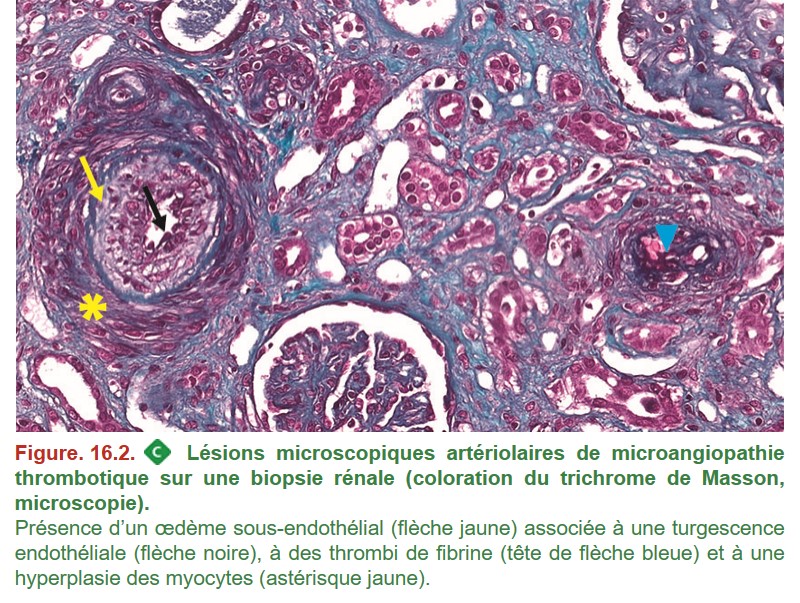{kind=link}
– turgescence des cellules endothéliales ;
– œdème sous-endothélial (au niveau glomérulaire on parle d’espaces clairs sous-endothéliaux) ;
– thrombi de fibrine et/ou de plaquettes ;
– hyperplasie concentrique des myocytes artériolaires (aspect en « bulbe d’oignon ») ;
– aspect ischémique ou congestif des glomérules ;
– mésangiolyse.
IF : thrombi de fibrine mis en évidence par l’anticorps antifibrinogène.
IV. Maladie des emboles de cristaux de cholestérol
• MO (Figure. 16.3) :
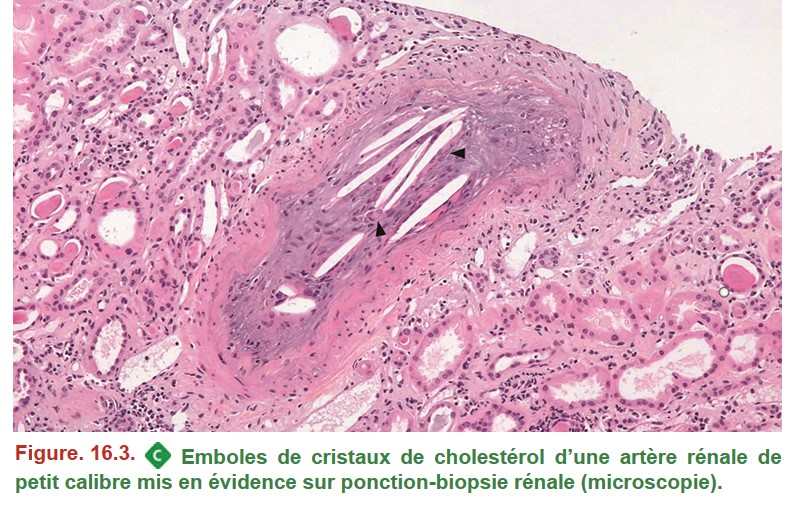{kind=link}
– cristaux de cholestérol artériolaires et/ou artériels ;
– réaction inflammatoire avec cellules géantes multinucléées au contact des cristaux.
Les cristaux apparaissent sous la forme de structures allongées optiquement vides (le cholestérol étant dissous lors des étapes de déshydratation des tissus) obstruant la lumière artérielle. Des cellules géantes multinucléées s’observent à leur contact (têtes de flèches). La présence des cristaux suscite un épaississement de l’intima participant à l’obstruction de la lumière.
Points clés
La néphroangiosclérose « bénigne » correspond au retentissement rénal de l’hypertension artérielle, entraînant l’apparition d’une insuffisance rénale chronique. La biopsie rénale n’est pas indispensable pour le diagnostic.
La microangiopathie thrombotique peut avoir de nombreuses causes ayant pour point commun une lésion de l’endothélium, provoquant une agrégation plaquettaire et la formation de thrombi fibrinoplaquettaires. Chez l’enfant, la biopsie rénale n’est pas nécessaire au diagnostic lorsque le tableau clinique est typique.
La maladie des emboles de cristaux de cholestérol correspond à la migration des cristaux de cholestérol par rupture de plaques athéromateuses, le plus souvent déclenchée par un geste vasculaire ou la prise de médicaments anticoagulants/antiagrégants. Le diagnostic peut être fait par examen du fond d’œil, biopsie cutanée ou biopsie rénale.
Item 195 – Artérite à cellules géantes
Auteur : Jean-Baptiste Gibier
I. Prérequis : la paroi artérielle normale
II. Introduction
III. Artérite à cellules géantes
IV. Maladie de Takayasu
Hiérarchisation des connaissances Tableau 4
{kind=link}
I. Prérequis : la paroi artérielle normale
La paroi artérielle est constituée de trois tuniques (Figure. 17.1) :
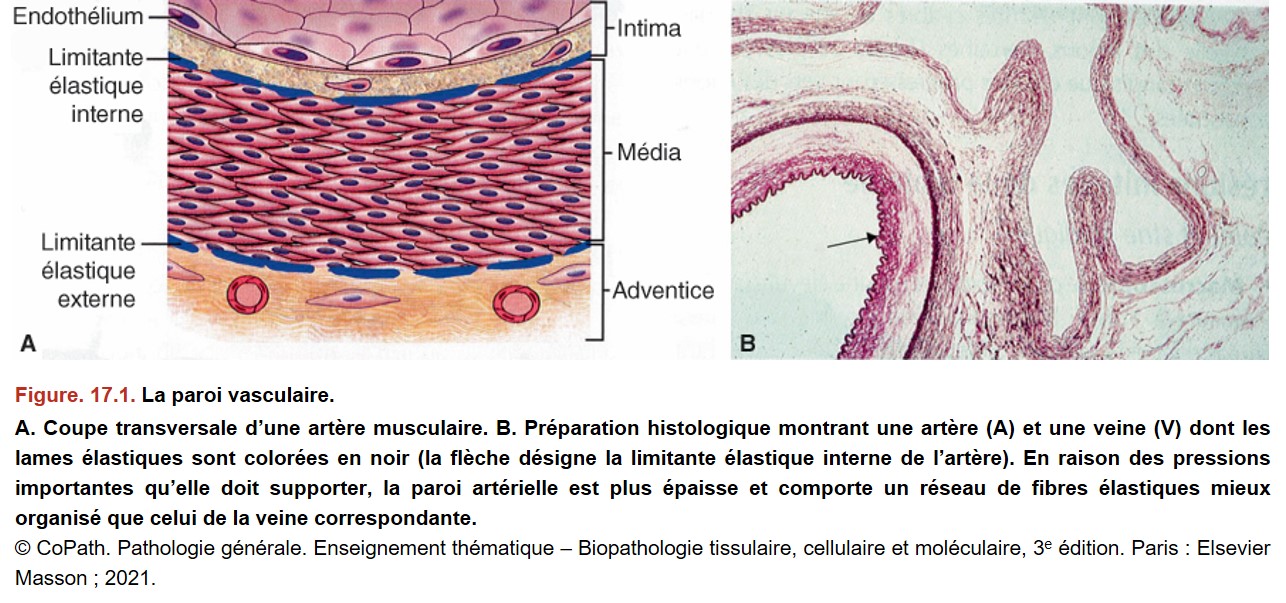{kind=link}
• l’intima : endothélium et zone sous-endothéliale ;
• séparée par la limitante élastique interne de la média : tunique d’épaisseur variable en fonction du calibre du vaisseau, constituée de cellules musculaires lisses et de fibres élastiques ;
• l’adventice, tissu conjonctif (renfermant notamment les vasa vasorum pour les artères de gros calibre), séparée de la média par la limitante élastique externe.
II. Introduction
III. Artérite à cellules géantes
L’artérite à cellules géantes est la vascularite systémique la plus fréquente après 50 ans. Elle atteint préférentiellement les artères de gros calibre (aorte et ses branches de division), notamment les branches de l’artère carotide externe (artères temporales et occipitales).
A. Manifestations cliniques
Le plus fréquemment, l’atteinte est limitée ou prédomine au niveau céphalique mais dans 10 % des cas, elle peut intéresser d’autres territoires. Les signes les plus fréquemment observés sont les suivants :
• signes généraux : fièvre (hyperthermie/fièvre), asthénie, anorexie, amaigrissement ;
• signes rhumatismaux : raideurs et douleurs articulaires d’horaire inflammatoire des ceintures pelvienne et scapulaire (PPR [pseudo-polyarthrite rhizomélique] associée) ;
• signes céphaliques et ophtalmiques : céphalées, hyperesthésie du cuir chevelu, claudication de la mâchoire, diminution du pouls temporal, neuropathie ischémique antérieure aiguë ;
• signes vasculaires : aortite, claudication d’un membre, souffle vasculaire, AVC (accident vasculaire cérébral).
B. Diagnostic
• lorsque la suspicion clinique est forte, elle ne doit pas retarder le début du traitement qui est une urgence ;
• elle doit être unilatérale, guidée par la clinique (et parfois par l’échographie) ;
• elle doit être de bonne taille, ≥ 5-10 mm de long, en raison du caractère segmentaire et focal des lésions ;
• elle a une sensibilité diagnostique de l’ordre de 70 % sa négativité n’exclut pas le diagnostic.
• en totalité le prélèvement (inclusion en totalité) ;
• sur de multiples niveaux (car lésions segmentaires et focales) ;
• avec une coloration spéciale soulignant les fibres élastiques (pour rechercher une rupture de la limitante élastique interne).
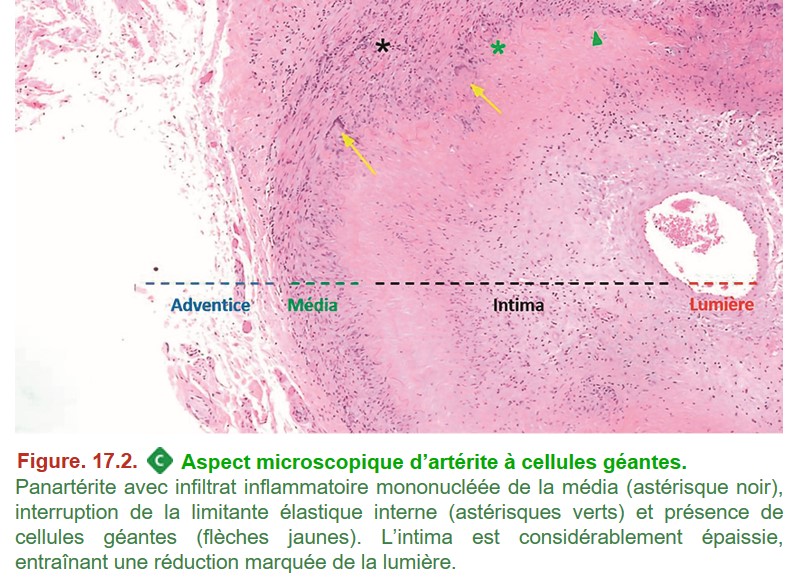
C. Lésions histologiques
{kind=link}
• inflammation mononucléée : lymphocytes, macrophages ;
• la présence de cellules géantes multinucléées (inconstantes) ;
• rupture de la limitante élastique interne (qui sépare la média de l’intima) ;
• épaississement (synonyme : hyperplasie) de l’intima responsable d’une sténose ou d’une occlusion du vaisseau ;
• absence de nécrose fibrinoïde.
Remarque : Bien que les cellules géantes soient inconstantes en histologie, le terme d’artérite à cellules géantes est retenu actuellement.
IV. Maladie de Takayasu
La maladie de Takayasu est une vascularite des vaisseaux de gros calibre. C’est une maladie très rare qui survient préférentiellement chez les femmes (sex ratio : 9 femmes pour 1 homme) de moins de 50 ans.
A. Manifestations cliniques
Une phase dite pré-occlusive est parfois observée avec notamment des signes généraux, des arthralgies, des épanchements pleuraux ou péricardiques et des douleurs sur les trajets des gros vaisseaux, en particulier la carotidodynie. Elle est suivie d’une phase occlusive qui est la conséquence de la sténose, l’occlusion ou l’anévrisme des artères. Au cours de cette phase, les manifestations dépendent donc du niveau de l’atteinte artérielle et sont caractérisées par : une claudication des membres (claudication intermittente d’un membre), des souffles vasculaires (découverte d’un souffle vasculaire), l’abolition d’un pouls, une HTA rénovasculaire, etc.
B. Diagnostic
L’imagerie vasculaire occupe une place primordiale dans le diagnostic et le suivi de la maladie. Les prélèvements biopsiques ne sont pas souvent réalisés.
C. Lésions histologiques
L’aspect histologique est très proche, voire superposable à celui observé dans l’artérite à cellules géantes.
Points clés
• Les vascularites sont caractérisées microscopiquement par une inflammation de la paroi vasculaire (artère et/ou veine).
• Concernant l’artérite à cellules géantes :
– c’est une vascularite systémique qui atteint les artères de gros et moyen calibre ;
– la biopsie de l’artère temporale est l’examen diagnostique clé ;
– la biopsie ne doit pas retarder le début du traitement. Elle doit être unilatérale, guidée par la clinique et de bonne taille (≥ 5-10 mm).
• Concernant l’examen anatomopathologique de la biopsie d’artère temporale :
– les lésions typiques sont une panartérite avec rupture de la limitante élastique interne et épaississement de l’intima, sans nécrose fibrinoïde ;
– la présence de cellules géantes est caractéristique mais inconstante.
Item 193 – Connaître les principaux types de vascularite systémique, les organes cibles, les outils diagnostiques et les moyens thérapeutiques
Auteur : Jean-Baptiste Gibier
I. Introduction
II. Manifestations cliniques des vascularites
III. Place de l’analyse histologique dans les vascularites de moyen calibre
IV. Place de l’analyse histologique dans les vascularites de petit calibre
Hiérarchisation des connaissances – Tableau 5
{kind=link}
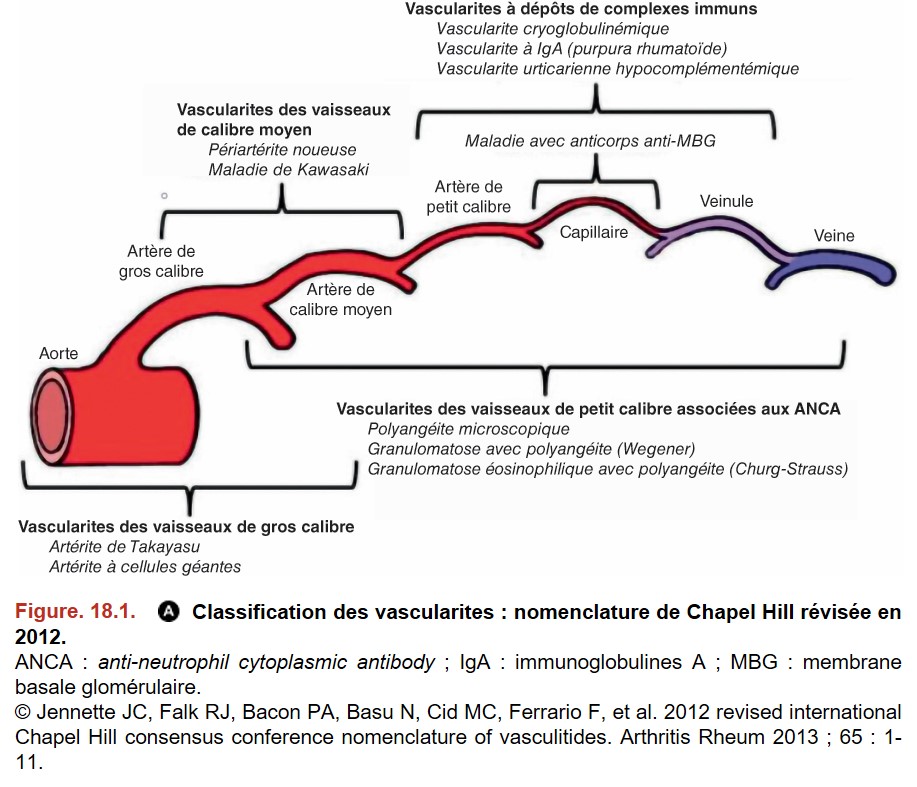
I. Introduction
{kind=link}
• vascularites des vaisseaux de gros calibre = aorte et ses branches de division :
•
•
II. Manifestations cliniques des vascularites
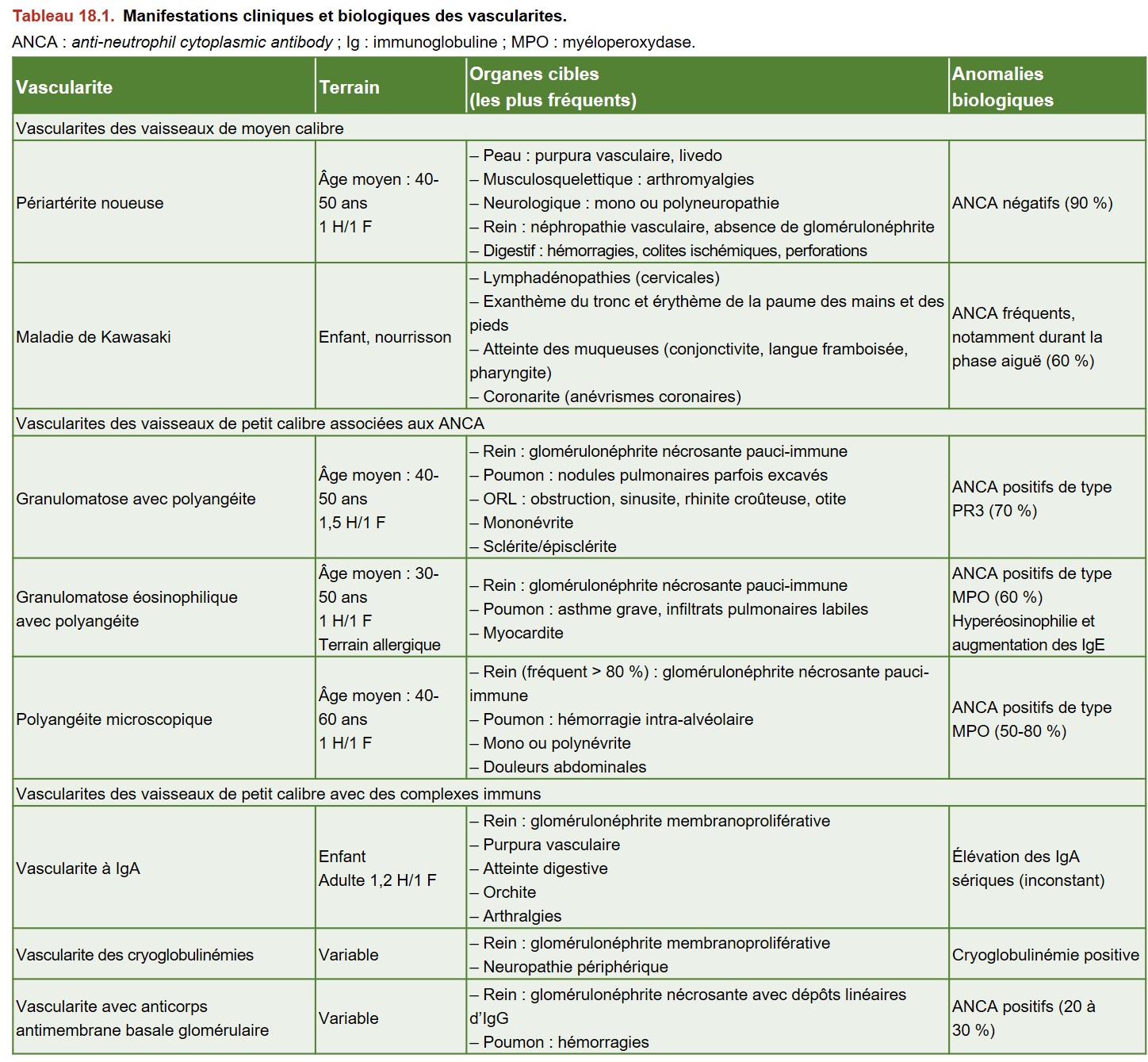
Les présentations cliniques de vascularite sont polymorphes et dépendent principalement du type de vaisseaux touchés. Les principales caractéristiques sont résumées dans le Tableau 18.1.
{kind=link}
Certains signes cliniques sont largement partagés par les vascularites de petit et moyen calibre, notamment le purpura vasculaire.
III. Place de l’analyse histologique dans les vascularites de moyen calibre
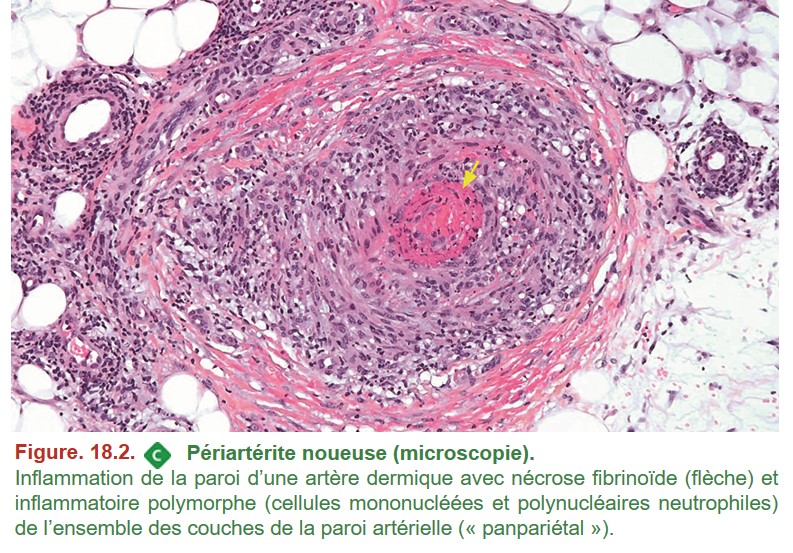
A. Périartérite noueuse
La réalisation technique de la biopsie cutanée est particulièrement importante pour porter le diagnostic de PAN car une biopsie trop superficielle est non contributive (préférer une biopsie fusiforme plutôt qu’une biopsie au punch).
{kind=link}
• une nécrose fibrinoïde de la média artérielle,
•
Attention :
B. Maladie de Kawasaki
L’analyse histologique n’est pas indiquée pour faire le diagnostic de la maladie.
IV. Place de l’analyse histologique dans les vascularites de petit calibre
A. Vascularites des petits vaisseaux associées aux ANCA
Trois entités sont regroupées dans la famille des vascularites des petits vaisseaux (artères de petit calibre, artérioles, veinules, capillaires) associées aux ANCA :
• granulomatose avec polyangéite (GPA, anciennement maladie de Wegener) ;
• polyangéite microscopique (PAM) ;
• granulomatose éosinophilique avec polyangéite (GEPA, anciennement syndrome de Churg et Strauss).
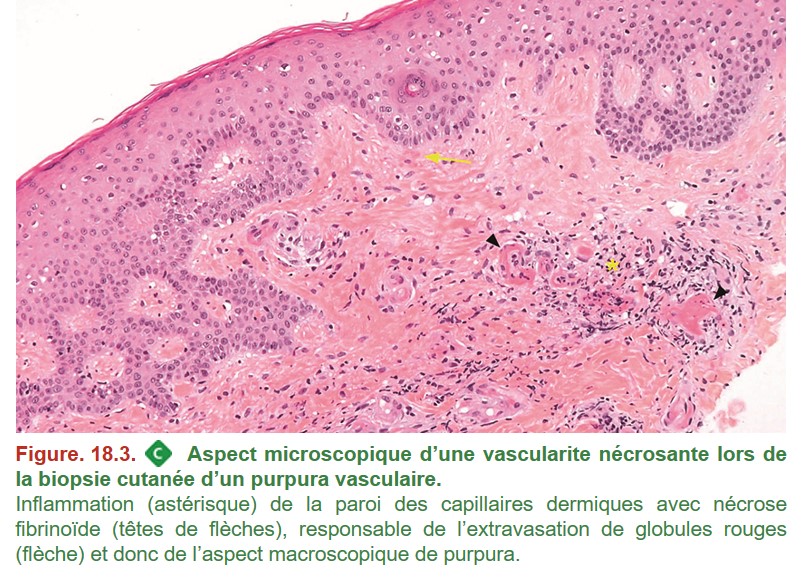
Les organes habituellement biopsiés sont le rein, le plus souvent dans un contexte de syndrome de glomérulonéphrite rapidement progressive et la peau en cas de purpura (Figure. 18.3).
{kind=link}
Plus rarement, en cas d’atteinte de la sphère ORL et de suspicion de granulomatose avec polyangéite, des biopsies sinusiennes peuvent être réalisées. Les biopsies neuromusculaires peuvent parfois être discutées en cas de neuropathie périphérique.
GPA, PAM et GEPA sont, comme la PAN, des vascularites nécrosantes. Sur le plan anatomopathologique, la présence d’une nécrose fibrinoïde de la paroi des vaisseaux atteints est donc commune à ces trois maladies.
La présence de granulomes épithélioïdes oriente vers un diagnostic de GPA ou de GEPA et un infiltrat inflammatoire riche en éosinophiles vers le diagnostic de GEPA.
Un examen en IF à partir de tissu congelé (dont les modalités techniques sont décrites dans l’item 261 Néphropathie glomérulaire) peut être nécessaire pour distinguer les deux grands groupes de vascularites des petits vaisseaux :
• en cas de vascularite associée aux ANCA, l’IF ne met pas en évidence de dépôts d’immunoglobulines ou de complément au niveau des lésions vasculaires (d’où le terme de « vascularite pauci-immune ») ;
• à l’inverse, des dépôts s’observent en cas de vascularite des petits vaisseaux avec dépôts de complexes immuns.
B. Vascularites des petits vaisseaux avec dépôts de complexes immuns
Quatre entités font partie de la famille des vascularites des petits vaisseaux avec dépôts de complexes immuns (= immunoglobulines + fractions du complément) :
• vascularite cryoglobulinémique ;
• vascularite à IgA (= purpura rhumatoïde) ;
• vascularite urticarienne hypocomplémentémique (non abordée dans le Tableau 18.1) ;
{kind=link}
• vascularite à anticorps antimembrane basale glomérulaire. La plupart de ces entités sont abordées dans l’item 261 Néphropathie glomérulaire
Comme pour les vascularites associées aux ANCA, les biopsies intéressent surtout le rein et la peau.
Les lésions artérielles sont superposables à celles décrites au cours de la PAN et des vascularites des petits vaisseaux associées aux ANCA. En revanche, l’atteinte glomérulaire au cours des vascularites cryoglobulinémiques et des vascularites à IgA se distingue par la présence d’une prolifération endocapillaire (celle-ci étant absente au cours de l’atteinte glomérulaire des vascularites associées aux ANCA). Cette dernière correspond à la réaction immunitaire locale secondaire à la présence des dépôts de complexes immuns.
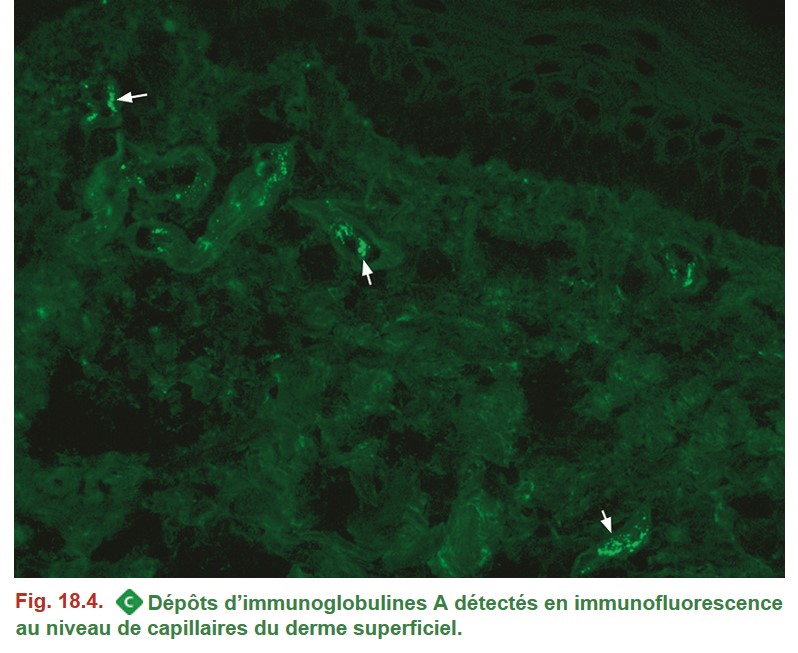
L’examen en IF apporte des informations importantes, en mettant en évidence la présence de dépôts immuns constitués d’immunoglobulines et de complément :
• dépôts granuleux d’immunoglobulines de complément au cours des vascularites cryoglobulinémiques. La constitution des dépôts varie en fonction de la nature de la cryoglobuline ;
• dépôts granuleux d’IgA au cours des vascularites à IgA (Figure. 18.4) ;
{kind=link}
• dépôts linéaires d’IgG le long de la membrane basale glomérulaire au cours des vascularites (ou glomérulonéphrites) à anticorps antimembrane basale glomérulaire.
Points clés
• Le diagnostic repose notamment sur la biopsie d’un organe atteint.
• Deux vascularites intéressent des vaisseaux de moyen calibre :
– la PAN, vascularite nécrosante avec inflammation et nécrose fibrinoïde de la paroi artérielle. Les biopsies cutanées et neuromusculaires dirigées ont la meilleure rentabilité pour le diagnostic ;
– la maladie de Kawasaki, artérite touchant les artères de moyen et parfois petit calibre. L’examen anatomopathologique n’est pas utile au diagnostic.
• S’agissant des vascularites des petits vaisseaux, également caractérisées par une nécrose vasculaire, l’examen en immunofluorescence d’une biopsie permet de distinguer le groupe des vascularites à ANCA (« pauci-immunes » = pas de dépôts) de celui des vascularites avec dépôts de complexes immuns.